About
|
Associate professor, Doctor of the Hungarian Academy of Sciences Head of MTA-SZTE Lendület Computational Reaction Dynamics Research Group Department of Physical Chemistry and Materials Science University of Szeged Rerrich Béla tér 1. H-6720 Szeged, Hungary Office: BE-417, Phone: +36-62-34-3742 E-mail: gczako at chem.u-szeged.hu |
Curriculum vitae
Date of Birth: January 1, 1981
Place of Birth: Szentes, Hungary
Degrees and positions
| 2022 | Visiting Professor, Department of Chemistry and Chemical Biology, University of New Mexico, Albuquerque, USA (3 months) |
| 2020 - | Leader of the Theoretical Chemistry Program, Doctoral School of Chemistry, University of Szeged, Szeged, Hungary |
| 2019 - | Associate Professor, Department of Physical Chemistry and Materials Science, University of Szeged, Szeged, Hungary |
| 2018 | Habilitation, University of Szeged, Szeged, Hungary |
| 2017 | Doctor of the Hungarian Academy of Sciences |
| 2015 - | Head of the Computational Reaction Dynamics Research Group, University of Szeged, Szeged, Hungary |
| 2015 - 2019 | Assistant Professor, Department of Physical Chemistry and Materials Science, University of Szeged, Szeged, Hungary |
| 2011 - 2015 | Research Associate, Institute of Chemistry, Eötvös University, Budapest, Hungary |
| 2008 - 2011 | Postdoctoral Fellow, Emory University, Atlanta, USA, in the group of Prof. Joel M. Bowman |
| 2007 | Ph.D. in Theoretical Chemistry, Eötvös University, Budapest, Hungary, Advisors: Prof. Attila G. Császár and Dr. Viktor Szalay |
| 2004 | M.Sc. in Chemistry, Eötvös University, Budapest, Hungary |
Grants and fellowships
| 2024 - 2029 | Momentum Program, Searching for quantum effects in the dynamics of polyatomic chemical reactions (Principal Investigator) |
| 2024 - 2027 | NKFIH K-146759, Automated modeling of chemical reaction dynamics (Principal Investigator) |
| 2022 - 2025 | TKP2021-NVA-19, Materials science and photonics (Participant) |
| 2019 - 2024 | Momentum Program, Computer-animated atomic-resolution movies revealing the dynamics and novel mechanisms of chemical reactions (Principal Investigator) |
| 2018 - 2021 | 20391-3/2018/FEKUSTRAT, Dynamics and mechanisms of fundamental gas-phase and surface reactions (Principal Investigator) |
| 2017 - 2021 | NKFIH K-125317, Understanding and controlling the atomic-level dynamics of chemical reactions: From fundamental processes to small biosystems (Principal Investigator) |
| 2015 - 2017 | OTKA PD-111900, Dynamics of polyatomic chemical reactions (Principal Investigator) |
| 2014 - 2017 | MTA-Bolyai, Atomic-level dynamics of chemical reactions (Principal Investigator) |
| 2013 - 2014 | A1-MZPD-12-0138, Dynamics of fundamental bimolecular chemical reactions (Principal Investigator) |
| 2012 - 2015 | MTA-Bolyai, Dynamics of fundamental bimolecular chemical reactions (Principal Investigator) |
| 2012 - 2014 | OTKA NN-81072-91234, Tunneling in novel hydroxycarbenes (Participant) |
| 2011 - 2014 | OTKA NK-83583, The fourth age of quantum chemistry (Participant) |
Academic honors and awards
| 2022 | Publication of the Year Prize, University of Szeged |
| 2020 | Young Researcher of the Year, University of Szeged |
| 2018 | Science Prize of the Faculty of Science and Informatics, University of Szeged |
| 2018 | Bolyai Plaquette of the Hungarian Academy of Sciences |
| 2015 | Top Reviewer for The Journal of Chemical Physics 2014 |
| 2014 | Bolyai Research Fellowship of the Hungarian Academy of Sciences (36 months) |
| 2013 | Magyary Fellowship (16 months) |
| 2012 | Junior Prima Prize in the Hungarian Science Category |
| 2012 | Bolyai Research Fellowship of the Hungarian Academy of Sciences (4 months) |
| 2012 | Junior Polányi Prize (best physical chemist in Hungary under age 35) |
| 2007 | Supervisor Award (National Competition of Research Students: II. place) |
| 2006 | Supervisor Award (Eötvös Competition of Research Students: III. place) |
| 2004 - 2007 | Hungarian State Doctoral Fellowship |
| 2004 | Excellent Student of the Faculty of Science of Eötvös University |
| 2003 - 2004 | Scholarship of the Republic of Hungary |
| 2003 | National Competition of Research Students: II. place |
| 2002 | Eötvös Competition of Research Students: I. place |
| 2000 | Young Talent of Szeged |
| 1999 - 2000 | Prime Minister's Scholarship |
| 1999 | Albert Szent-Györgyi Medal of the Hungarian Chemical Society and the Hungarian Society of Science |
| 1999 | Silver Medal in the 31st International Chemistry Olympiad, Bangkok, Thailand |
Visits
| 2022 Summer | University of New Mexico, Albuquerque, New Mexico USA (3 months) |
| 2006 Winter | Center for Computational Chemistry, Athens, Georgia USA (2 months) |
| 2004 Fall | Center for Computational Chemistry, Athens, Georgia USA (2 months) |
| 2003 Fall | Center for Computational Chemistry, Athens, Georgia USA (3 months) |
Professional activities
| Grant proposals |
| National Research, Development and Innovation Office (NKFIH) |
| Hungarian Scientific Research Fund (OTKA) |
| Hungarian Academy of Sciences |
| Austrian Science Fund |
| Czech Science Foundation |
| National Science Centre Poland |
| U.S. Department of Energy (DOE) Office of Science |
| Journal articles (about 30-40 reviews / year) |
| Science, Nature Chemistry, Nature Communications, Journal of the American Chemical Society, Angewandte Chemie, Chemical Science, Physical Chemistry Chemical Physics, Journal of Chemical Physics ["Top 20 JCP Reviewer (2014)" recognition], Journal of Physical Chemistry, Molecular Physics, International Journal of Quantum Chemistry, etc. |
| Head of the Chemometry and Molecular Modeling (KeMoMo) Working Group |
| Material and Molecular Structure Working Group of the Hungarian Academy of Sciences |
| Reaction Kinetics and Photochemistry Working Group of the Hungarian Academy of Sciences |
| Hungarian Chemical Society |
| Public body of the Hungarian Academy of Sciences |
| Physical Chemistry Scientific Committee of the Hungarian Academy of Sciences |
Research









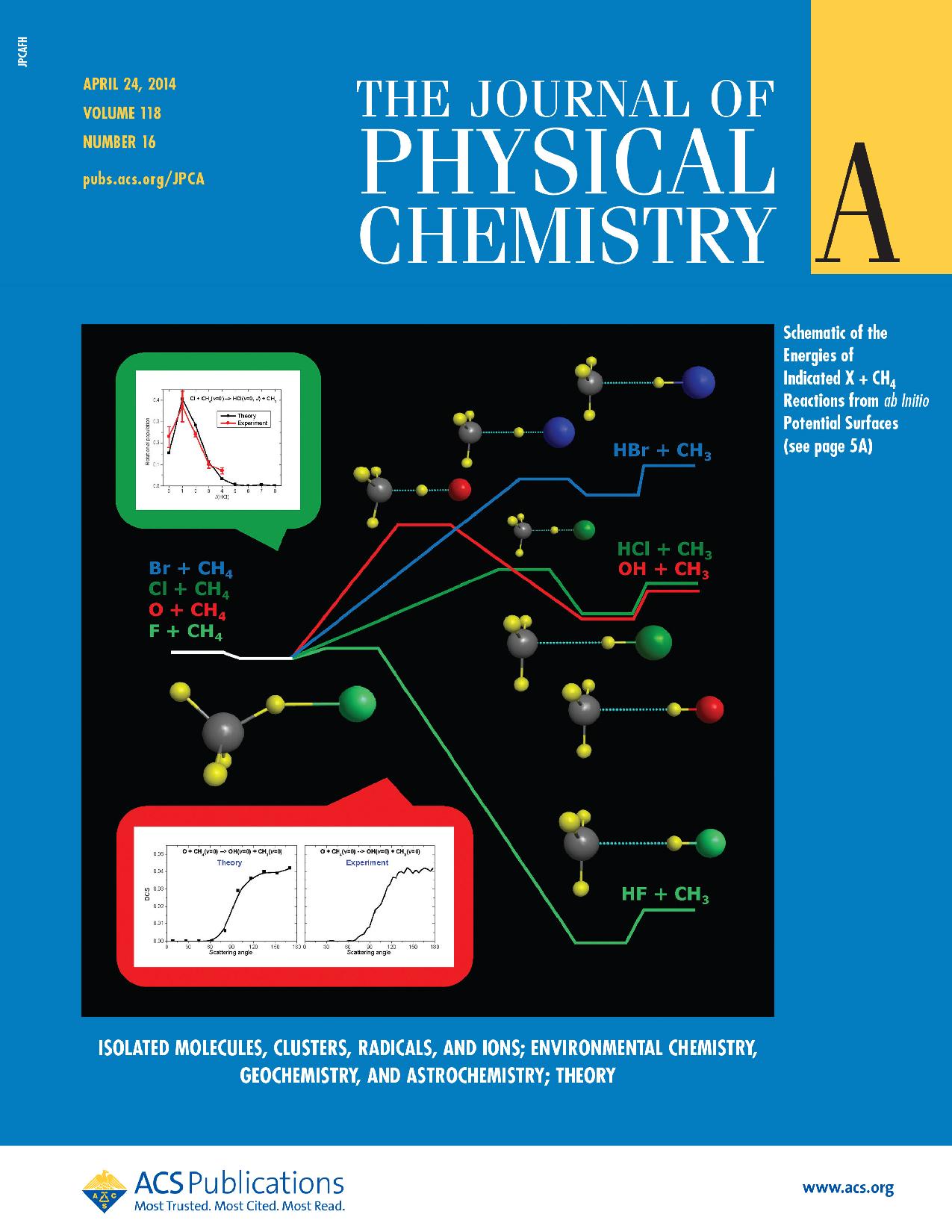


My early research focused on method developments for fully-anharmonic rotational-vibrational computations for small and medium-size molecules. In 2008 I started to work on the field of chemical reaction dynamics. We developed analytical ab initio potential energy surfaces for several atom plus methane and SN2 reactions and studied their dynamics with quasi-classical and/or quantum methods. Some of these studies were reported in high-profile multidisciplinary and chemistry journals such as Science, PNAS, Nature Chemistry, Nature Communications, JACS, and Chemical Science. We are currently extending our dynamics studies toward more complex systems from 7-10-atomic reactions to small biosystems.
Potential energy surface developments
- ROBOSURFER: A program package for automated PES development
- ManyHF: A pragmatic automated method of finding lower-energy Hartree−Fock solutions for PES development
- Atom/radical-molecule reactions: F + CH4, O + CH4, Cl + CH4, Br + CH4, F + C2H6, Cl + C2H6, OH + C2H6, HBr + C2H5, HI + C2H5, Cl + CH3NH2, CH2OO + NH3, F + CH3NH2, CH2OO + SO2, and Cl + CH3CN
- Ion-molecule reactions: F− + CH3F, F− + CH3Cl, F− + CH3Br, F− + CH3I, OH− + CH3F, OH− + CH3I, F− + NH2Cl, F− + CH3CH2Cl, NH2− + CH3I, Cl− + CH3I, F− + SiH3Cl, F− + PH2Cl, OH− + CH3CH2Cl, and F− + SiH3I
- Complexes: CH4.F− and CH4.Ar
Method developments
Applications
Development of full-dimensional ab initio PESs for the following systems:Reaction and molecular dynamics computations
- Mode-specific quasi-classical polyatomic product analysis method
- Gaussian binning (1GB) for polyatomic molecules
- Zero-point energy constrained quasi-classical trajectory method and its applications to small water clusters
- Modifications of the rules of Nobel-laureate John Polanyi for polyatomic systems
- Introducing the notion of rotational mode specificity
- Mapping the angle-dependent barrier for the Cl + CHD3 reaction
- Revealing a double-inversion mechanism for SN2 reactions
- Uncovering unexpected leaving group effects for SN2 reactions
- Deciphering front-side complex formation in SN2 reactions via dynamics mapping
- Achieving unprecedented agreement with experiment on SN2 reaction dynamics
- Disentangling substitution (SN2) and elimination (E2) reaction pathways
Method developments
Dynamics of the reactions of atoms with methane
Dynamics of SN2 reactions
First-principles rotational-vibrational spectroscopy
-
Developments and applications of discrete variable representation-based methods and computer codes for computing ro-vibrational energy levels of triatomic and medium-size molecules
Non-Born-Oppenheimer computations
-
Adiabatic Jacobi corrections for H2+ and HD+
Publications
|
Number of publications:153 Total citations:5372(6377) Cumulative impact factor:655 H-index:43(46) Web of Science(Google Scholar) (As of July 15, 2025) |
V. Szalay, G. Czakó, Á. Nagy, T. Furtenbacher, and A. G. Császár
On one-dimensional discrete variable representations with general basis functions
J. Chem. Phys. 119, 10512 (2003) PDF
G. Czakó, T. Furtenbacher, A. G. Császár, and V. Szalay
Variational vibrational calculations using high-order anharmonic force fields
Mol. Phys. 102, 2411 (2004) PDF
G. Czakó, V. Szalay, A. G. Császár, and T. Furtenbacher
Treating singularities present in the Sutcliffe-Tennyson vibrational Hamiltonian in orthogonal internal coordinates
J. Chem. Phys. 122, 024101 (2005) PDF
A. G. Császár, G. Czakó, T. Furtenbacher, J. Tennyson, V. Szalay, S. V. Shirin, N. F. Zobov, and O. L. Polyansky
On equilibrium structures of the water molecule
J. Chem. Phys. 122, 214305 (2005) PDF
G. Tarczay, T. A. Miller, G. Czakó, and A. G. Császár
Accurate ab initio determination of spectroscopic and thermochemical properties of mono- and dichlorocarbenes
Phys. Chem. Chem. Phys. 7, 2881 (2005) PDF
T. Furtenbacher, G. Czakó, B. T. Sutcliffe, A. G. Császár, and V. Szalay
The methylene saga continues: stretching fundamentals and zero-point energy of X 3B1 CH2
J. Mol. Struct. 780-781, 283 (2006) PDF
G. Czakó, V. Szalay, and A. G. Császár
Finite basis representations with nondirect-product basis functions having structure similar to that of spherical harmonics
J. Chem. Phys. 124, 014110 (2006) PDF
A. G. Császár, T. Furtenbacher, and G. Czakó
The greenhouse effect on Earth and the complete spectroscopy of water
Magy. Kém. Foly. 112, 123 (2006) (in Hungarian) PDF
G. Czakó, A. G. Császár, V. Szalay, and B. T. Sutcliffe
Adiabatic Jacobi corrections for H2+-like systems
J. Chem. Phys. 126, 024102 (2007) PDF
A. G. Császár, G. Czakó, T. Furtenbacher, and E. Mátyus
An active database approach to complete rotational-vibrational spectra of small molecules
Ann. Rep. Comp. Chem. 3, 155 (2007) PDF
G. Czakó, T. Furtenbacher, P. Barletta, A. G. Császár, V. Szalay, and B. T. Sutcliffe
Use of a nondirect-product basis for treating singularities in triatomic rotational-vibrational calculations
Phys. Chem. Chem. Phys. 9, 3407 (2007) PDF
E. Mátyus, G. Czakó, B. T. Sutcliffe, and A. G. Császár
Vibrational energy levels with arbitrary potentials using the Eckart-Watson Hamiltonians and the discrete variable representation
J. Chem. Phys. 127, 084102 (2007) PDF
G. Czakó, E. Mátyus, A. C. Simmonett, A. G. Császár, H. F. Schaefer III, and W. D. Allen
Anchoring the absolute proton affinity scale
J. Chem. Theory Comput. 4, 1220 (2008) PDF
G. Czakó, B. J. Braams, and J. M. Bowman
Accurate ab initio structure, dissociation energy, and vibrational spectroscopy of the F−_CH4 anion complex
J. Phys. Chem. A 112, 7466 (2008) PDF
G. Tarczay, T. A. Miller, G. Czakó, and A. G. Császár
Additions and corrections to "Accurate ab initio determination of spectroscopic and thermochemical properties of mono- and dichlorocarbenes"
Phys. Chem. Chem. Phys. 10, 7324 (2008) PDF
T. Szidarovszky, G. Czakó, and A. G. Császár
Conformers of gaseous threonine
Mol. Phys. 107, 761 (2009) PDF
G. Czakó, B. C. Shepler, B. J. Braams, and J. M. Bowman
Accurate ab initio potential energy surface, dynamics, and thermochemistry of the F + CH4 __> HF + CH3 reaction
J. Chem. Phys. 130, 084301 (2009) PDF Highlighted by JCP: Editors' Choice for 2009: Gas phase dynamics
G. Czakó, B. Nagy, G. Tasi, Á. Somogyi, J. Simunek, J. Noga, B. J. Braams, J. M. Bowman, and A. G. Császár
Proton affinity and enthalpy of formation of formaldehyde
Int. J. Quant. Chem. 109, 2393 (2009) PDF
E. Mátyus, G. Czakó, and A. G. Császár
Toward black-box-type full- and reduced-dimensional variational (ro)vibrational computations
J. Chem. Phys. 130, 134112 (2009) PDF
C. Fábri, G. Czakó, G. Tasi, and A. G. Császár
Adiabatic Jacobi corrections on the vibrational energy levels of H2+ isotopologues
J. Chem. Phys. 130, 134314 (2009) PDF
G. Czakó, E. Mátyus, and A. G. Császár
Bridging theory with experiment: a benchmark study of thermally averaged structural and effective spectroscopic parameters of the water molecule
J. Phys. Chem. A 113, 11665 (2009) PDF
G. Czakó and J. M. Bowman
CH stretching excitation steers the F atom to the CD bond in the F + CHD3 reaction
J. Am. Chem. Soc. 131, 17534 (2009) PDF Highlighted by Science, Editors' Choice: Fluorine diverted
G. Czakó and J. M. Bowman
Quasiclassical trajectory calculations of correlated product distributions for the F + CHD3(v1= 0, 1) reactions using an ab initio potential energy surface
J. Chem. Phys. 131, 244302 (2009) PDF
E. Mátyus, C. Fábri, T. Szidarovszky, G. Czakó, W. D. Allen, and A. G. Császár
Assigning quantum labels to variationally computed rotational-vibrational eigenstates of polyatomic molecules
J. Chem. Phys. 133, 034113 (2010) PDF
T. Szidarovszky, A. G. Császár, and G. Czakó
On the efficiency of treating singularities in triatomic variational vibrational computations. The vibrational states of H3+ up to dissociation
Phys. Chem. Chem. Phys. 12, 8373 (2010) PDF
G. Czakó, A. L. Kaledin, and J. M. Bowman
A practical method to avoid zero-point leak in molecular dynamics calculations: Application to the water dimer
J. Chem. Phys. 132, 164103 (2010) PDF
J. M. Bowman, B. J. Braams, S. Carter, C. Chen, G. Czakó, B. Fu, X. Huang, E. Kamarchik, A. R. Sharma, B. C. Shepler, Y. Wang, and Z. Xie
Ab initio-based potential energy surfaces for complex molecules and molecular complexes
J. Phys. Chem. Lett. 1, 1866 (2010) Perspective PDF
G. Czakó, Q. Shuai, K. Liu, and J. M. Bowman
Communication: Experimental and theoretical investigations of the effects of the reactant bending excitations in the F + CHD3 reaction
J. Chem. Phys. 133, 131101 (2010) PDF One of the Top 20 Most Downloaded JCP articles in October 2010
G. Czakó, A. L. Kaledin, and J. M. Bowman
Zero-point energy constrained quasiclassical, classical, and exact quantum simulations of isomerizations and radial distribution functions of the water trimer using an ab initio potential energy surface
Chem. Phys. Lett. 500, 217 (2010) PDF
G. Czakó and J. M. Bowman
An ab initio spin-orbit-corrected potential energy surface and dynamics for the F + CH4 and F + CHD3 reactions
Phys. Chem. Chem. Phys. 13, 8306 (2011) PDF
J. M. Bowman, G. Czakó, and B. Fu
High-dimensional ab initio potential energy surfaces for reaction dynamics calculations
Phys. Chem. Chem. Phys. 13, 8094 (2011) Perspective PDF
M. Cheng, Y. Feng, Y. Du, Q. Zhu, W. Zheng, G. Czakó, and J. M. Bowman
Communication: Probing the entrance channels of the X + CH4 __> HX + CH3 (X = F, Cl, Br, I) reactions via photodetachment of X-_CH4
J. Chem. Phys. 134, 191102 (2011) PDF
G. Czakó, Y. Wang, and J. M. Bowman
Communication: Quasiclassical trajectory calculations of correlated product-state distributions for the dissociation of (H2O)2 and (D2O)2
J. Chem. Phys. 135, 151102 (2011) PDF One of the Top 20 Most Downloaded JCP articles in October 2011
G. Czakó and J. M. Bowman
Dynamics of the reaction of methane with chlorine atom on an accurate potential energy surface
Science 334, 343 (2011) PDF Highlighted by ChemPhysChem: "Reaction Dynamics: Rules Change with Molecular Size"
A. G. Császár, C. Fábri, T. Szidarovszky, E. Mátyus, T. Furtenbacher, and G. Czakó
The fourth age of quantum chemistry: molecules in motion
Phys. Chem. Chem. Phys. 14, 1085 (2012) Perspective Cover PDF One of the Top 10 Most Downloaded PCCP articles in December 2011
G. Czakó and J. M. Bowman
Accurate ab initio potential energy surface, thermochemistry, and dynamics of the Cl(2P, 2P3/2) + CH4 __> HCl + CH3 and H + CH3Cl reactions
J. Chem. Phys. 136, 044307 (2012) PDF One of the Most Cited 2012 Articles published in JCP
V. Szalay, T. Szidarovszky, G. Czakó, and A. G. Császár
A paradox of grid-based representation techniques: accurate eigenvalues from inaccurate matrix elements
J. Math. Chem. 50, 636 (2012) PDF
B. Zhang, K. Liu, G. Czakó, and J. M. Bowman
Translational energy dependence of the Cl + CH4(vb= 0, 1) reactions: A joint crossed-beam and quasiclassical trajectory study
Mol. Phys. 110, 1617 (2012) PDF
G. Czakó and J. M. Bowman
Dynamics of the O(3P) + CHD3(vCH= 0, 1) reactions on an accurate ab initio potential energy surface
Proc. Natl. Acad. Sci. U.S.A. 109, 7997 (2012) PDF
I. Szabó, C. Fábri, G. Czakó, E. Mátyus, and A. G. Császár
Temperature-dependent, effective structures of the 14NH3 and 14ND3 molecules
J. Phys. Chem. A 116, 4356 (2012) PDF
A. G. Császár, G. Czakó, T. Furtenbacher, E. Mátyus, C. Fábri, T. Szidarovszky, I. Szabó, and J. Sarka
Molecular structure and dynamics
Magy. Kém. Foly. 118, 181 (2012) (in Hungarian) PDF
G. Czakó
Gaussian binning of the vibrational distributions for the Cl + CH4(v4/2=0,1) __> H + CH3Cl(n1n2n3n4n5n6) reactions
J. Phys. Chem. A 116, 7467 (2012) PDF
L. C. Ch'ng, A. K. Samanta, G. Czakó, J. M. Bowman, and H. Reisler
Experimental and theoretical investigations of energy transfer and hydrogen-bond breaking in the water dimer
J. Am. Chem. Soc. 134, 15430 (2012) PDF
Z. Zhang, Y. Zhou, D. H. Zhang, G. Czakó, and J. M. Bowman
Theoretical study of the validity of the Polanyi rules for the late-barrier Cl + CHD3 reaction
J. Phys. Chem. Lett. 3, 3416 (2012) PDF Highlighted by Science, Editors' Choice: Stretching the Polanyi Rules
R. Liu, M. Yang, G. Czakó, J. M. Bowman, J. Li, and H. Guo
Mode selectivity for a "central" barrier reaction: Eight-dimensional quantum studies of the O(3P) + CH4 __> OH + CH3 reaction on an ab initio potential energy surface
J. Phys. Chem. Lett. 3, 3776 (2012) PDF
C. Fábri, A. G. Császár, and G. Czakó
Reduced-dimensional quantum computations for the rotational-vibrational dynamics of F−_CH4 and F−_CH2D2
J. Phys. Chem. A 117, 6975 (2013) PDF
G. Czakó
Accurate ab initio potential energy surface, thermochemistry, and dynamics of the Br(2P, 2P3/2) + CH4 __> HBr + CH3 reaction
J. Chem. Phys. 138, 134301 (2013) PDF
D. Y. Wang and G. Czakó
Quantum dynamics study of the F + CH4 __> HF + CH3 reaction on an ab initio potential energy surface
J. Phys. Chem. A 117, 7124 (2013) PDF
G. Czakó, R. Liu, M. Yang, J. M. Bowman, and H. Guo
Quasiclassical trajectory studies of the O(3P) + CX4(vk=0,1) __> OX(v) + CX3(n1n2n3n4) [X = H and D] reactions on an ab initio potential energy surface
J. Phys. Chem. A 117, 6409 (2013) PDF
I. Szabó, A. G. Császár, and G. Czakó
Dynamics of the F− + CH3Cl __> Cl− + CH3F SN2 reaction on a chemically accurate potential energy surface
Chem. Sci. 4, 4362 (2013) Cover PDF One of the "HOT Chemical Science articles for October"
G. Czakó, I. Szabó, and H. Telekes
On the choice of the ab initio level of theory for potential energy surface developments
J. Phys. Chem. A 118, 646 (2014) PDF
G. Czakó and J. M. Bowman
Reaction dynamics of methane with F, O, Cl, and Br on ab initio potential energy surfaces
J. Phys. Chem. A 118, 2839 (2014) Feature Article Cover PDF
G. Czakó
Communication: Direct comparison between theory and experiment for correlated angular and product-state distributions of the ground-state and stretching-excited O(3P) + CH4 reactions
J. Chem. Phys. 140, 231102 (2014) PDF One of the Editors' Picks from The Journal of Chemical Physics
A. K. Samanta, G. Czakó, Y. Wang, J. S. Mancini, J. M. Bowman, and H. Reisler
Experimental and theoretical investigations of energy transfer and hydrogen-bond breaking in small water and HCl clusters
Acc. Chem. Res. 47, 2700 (2014) PDF
R. Liu, F. Wang, B. Jiang, G. Czakó, M. Yang, K. Liu, and H. Guo
Rotational mode specificity in the Cl + CHD3 __> HCl + CD3 reaction
J. Chem. Phys. 141, 074310 (2014) PDF
G. Czakó, A. G. Császár, and H. F. Schaefer III
Surprising quenching of the spin-orbit interaction significantly diminishes H2O...X [X = F, Cl, Br, I] dissociation energies
J. Phys. Chem. A 118, 11956 (2014) PDF
G. Czakó
Quasiclassical trajectory study of the rotational mode specificity in the O(3P) + CHD3(v1=0,1, JK) __> OH + CD3 reactions
J. Phys. Chem. A 118, 11683 (2014) PDF
I. Szabó and G. Czakó
Revealing a double-inversion mechanism for the F− + CH3Cl SN2 reaction
Nat. Commun. 6, 5972 (2015) PDF Highly Cited Paper (as of July/August 2015), Highlighted by National Geographic Hungary
B. Zhang, K. Liu, and G. Czakó
Correlated dynamics of the O(3P) + CHD3(v = 0) reaction: A joint crossed-beam and quasiclassical trajectory study
J. Phys. Chem. A 119, 7190 (2015) PDF
I. Szabó and G. Czakó
Double-inversion mechanisms of the X− + CH3Y [X,Y = F, Cl, Br, I] SN2 reactions
J. Phys. Chem. A 119, 3134 (2015) PDF
H. Wang, Y. Qiu, G. Czakó, and H. F. Schaefer III
Pathways for the OH + Cl2 __> HOCl + Cl and HOCl + Cl __> HCl + ClO reactions
J. Phys. Chem. A 119, 7802 (2015) PDF
I. Szabó, H. Telekes, and G. Czakó
Accurate ab initio potential energy surface, thermochemistry, and dynamics of the F− + CH3F SN2 and proton-abstraction reactions
J. Chem. Phys. 142, 244301 (2015) PDF
I. Szabó and G. Czakó
Rotational mode specificity in the F− + CH3Y [Y = F and Cl] SN2 reactions
J. Phys. Chem. A 119, 12231 (2015) PDF
M. Stei, E. Carrascosa, M. A. Kainz, A. H. Kelkar, J. Meyer, I. Szabó, G. Czakó, and R. Wester
Influence of the leaving group on the dynamics of a gas-phase SN2 reaction
Nat. Chem. 8, 151 (2016) PDF Highly Cited Paper (as of July/August 2016)
Y. Wang, H. Song, I. Szabó, G. Czakó, H. Guo, and M. Yang
Mode-specific SN2 reaction dynamics
J. Phys. Chem. Lett. 7, 3322 (2016) PDF
I. Szabó and G. Czakó
Mode-specific multi-channel dynamics of the F− + CHD2Cl reaction on a global ab initio potential energy surface
J. Chem. Phys. 145, 134303 (2016) PDF
B. Olasz, I. Szabó, and G. Czakó
High-level ab initio potential energy surface and dynamics of the F− + CH3I SN2 and proton-transfer reactions
Chem. Sci. 8, 3164 (2017) PDF
V. Tajti and G. Czakó
Benchmark ab initio characterization of the complex potential energy surface of the F− + CH3CH2Cl reaction
J. Phys. Chem. A 121, 2847 (2017) PDF
I. Szabó, B. Olasz, and G. Czakó
Deciphering front-side complex formation in SN2 reactions via dynamics mapping
J. Phys. Chem. Lett. 8, 2917 (2017) PDF
I. Szabó and G. Czakó
Benchmark ab initio characterization of the complex potential energy surface of the Cl− + CH3I reaction
J. Phys. Chem. A 121, 5748 (2017) PDF
H. Pan, F. Wang, G. Czakó, and K. Liu
Direct mapping of the angle-dependent barrier to reaction for Cl + CHD3 using polarized scattering data
Nat. Chem. 9, 1175 (2017) PDF
I. Szabó and G. Czakó
Dynamics and novel mechanisms of SN2 reactions on ab initio analytical potential energy surfaces
J. Phys. Chem. A 121, 9005 (2017) Feature Article Cover PDF
L. Krotos and G. Czakó
Does the Cl + CH4 __> H + CH3Cl reaction proceed via Walden inversion?
J. Phys. Chem. A 121, 9415 (2017) PDF
B. Hajdu and G. Czakó
Benchmark ab initio characterization of the complex potential energy surfaces of the X− + NH2Y [X,Y = F, Cl, Br, I] reactions
J. Phys. Chem. A 122, 1886 (2018) PDF
T. Győri, B. Olasz, G. Paragi, and G. Czakó
Effects of the level of electronic structure theory on the dynamics of the F− + CH3I reaction
J. Phys. Chem. A 122, 3353 (2018) PDF
S. Góger, P. Szabó, G. Czakó, and G. Lendvay
Flame inhibition chemistry: rate coefficients of the reactions of HBr with CH3 and OH radicals at high temperatures determined by quasiclassical trajectory calculations
Energy Fuels 32, 10100 (2018) PDF
M. Stei, E. Carrascosa, A. Dörfler, J. Meyer, B. Olasz, G. Czakó, A. Li, H. Guo, and R. Wester
Stretching vibration is spectator in nucleophilic substitution
Sci. Adv. 4, eaas9544 (2018) PDF
D. A. Tasi, Z. Fábián, and G. Czakó
Benchmark ab initio characterization of the inversion and retention pathways of the OH− + CH3Y [Y = F, Cl, Br, I] SN2 reactions
J. Phys. Chem. A 122, 5773 (2018) PDF
B. Olasz and G. Czakó
Mode-specific quasiclassical dynamics of the F− + CH3I SN2 and proton-transfer reactions
J. Phys. Chem. A 122, 8143 (2018) PDF
D. Papp, B. Gruber, and G. Czakó
Detailed benchmark ab initio mapping of the potential energy surfaces of the X + C2H6 [X = F, Cl, Br, I] reactions
Phys. Chem. Chem. Phys. 21, 396 (2019) PDF
B. Bastian, E. Carrascosa, A. Kaiser, J. Meyer, T. Michaelsen, G. Czakó, W. L. Hase, and R. Wester
Dynamics of proton transfer from ArH+ to CO
Int. J. Mass Spectrom. 438, 175 (2019) PDF
B. Olasz and G. Czakó
High-level-optimized stationary points for the F−(H2O) + CH3I system: Proposing a new water-induced double-inversion pathway
J. Phys. Chem. A 123, 454 (2019) PDF
B. Olasz and G. Czakó
Uncovering the role of the stationary points in the dynamics of the F− + CH3I reaction
Phys. Chem. Chem. Phys. 21, 1578 (2019) PDF
D. A. Tasi, Z. Fábián, and G. Czakó
Rethinking the X− + CH3Y [X = OH, SH, CN, NH2, PH2; Y = F, Cl, Br, I] SN2 reactions
Phys. Chem. Chem. Phys. 21, 7924 (2019) PDF
G. Czakó
Dynamics and mechanisms of fundamental chemical reactions
Magy. Kém. Foly. 125, 100 (2019) (in Hungarian) PDF
G. Czakó, T. Győri, B. Olasz, D. Papp, I. Szabó, V. Tajti, and D. A. Tasi
Benchmark ab initio and dynamical characterization of the stationary points of reactive atom + alkane and SN2 potential energy surfaces
Phys. Chem. Chem. Phys. 22, 4298 (2020) Perspective Cover PDF
T. Győri and G. Czakó
Automating the development of high-dimensional reactive potential energy surfaces with the ROBOSURFER program system
J. Chem. Theory Comput. 16, 51 (2020) PDF Highly Cited Paper (as of May/June 2020)
G. Avila, D. Papp, G. Czakó, and E. Mátyus
Exact quantum dynamics background of dispersion interactions: case study for CH4.Ar in full (12) dimensions
Phys. Chem. Chem. Phys. 22, 2792 (2020) PDF Selected as a 2020 HOT PCCP article
D. A. Tasi, T. Győri, and G. Czakó
On the development of a gold-standard potential energy surface for the OH− + CH3I reaction
Phys. Chem. Chem. Phys. 22, 3775 (2020) Communication PDF Selected as a 2020 HOT PCCP article
D. Papp, V. Tajti, T. Győri, and G. Czakó
Theory finally agrees with experiment for the dynamics of the Cl + C2H6 reaction
J. Phys. Chem. Lett. 11, 4762 (2020) PDF Publication of the Month (Chemical Sciences Section, Hungarian Academy of Sciences)
E. M. Orján, A. B. Nacsa, and G. Czakó
Conformers of dehydrogenated glycine isomers
J. Comput. Chem. 41, 2001 (2020) PDF
B. Gruber and G. Czakó
Benchmark ab initio characterization of the abstraction and substitution pathways of the OH + CH4/C2H6 reactions
Phys. Chem. Chem. Phys. 22, 14560 (2020) PDF
P. Papp, V. Tajti, and G. Czakó
Numerical separation of the front-side attack and double-inversion retention pathways of SN2 reactions
Chem. Phys. Lett. 755, 137780 (2020) PDF
D. Papp and G. Czakó
Full-dimensional MRCI-F12 potential energy surface and dynamics of the F(2P3/2) + C2H6 __> HF + C2H5 reaction
J. Chem. Phys. 153, 064305 (2020) PDF
P. Papp and G. Czakó
Rotational mode specificity in the F− + CH3I(v=0, JK) SN2 and proton-transfer reactions
J. Phys. Chem. A 124, 8943 (2020) PDF
G. Czakó, T. Győri, D. Papp, V. Tajti, and D. A. Tasi
First-principles reaction dynamics beyond six-atom systems
J. Phys. Chem. A 125, 2385 (2021) Perspective Cover PDF
D. Papp and G. Czakó
Facilitated inversion complicates the stereodynamics of an SN2 reaction at nitrogen center
Chem. Sci. 12, 5410 (2021) Cover PDF
A. B. Nacsa and G. Czakó
Benchmark ab initio proton affinity of glycine
Phys. Chem. Chem. Phys. 23, 9663 (2021) Cover PDF
T. Szűcs and G. Czakó
Benchmark ab initio stationary-point characterization of the complex potential energy surface of the multi-channel Cl + CH3NH2 reaction
Phys. Chem. Chem. Phys. 23, 10347 (2021) PDF
D. A. Tasi, C. Tokaji, and G. Czakó
A benchmark ab initio study of the complex potential energy surfaces of the OH− + CH3CH2Y [Y = F, Cl, Br, I] reactions
Phys. Chem. Chem. Phys. 23, 13526 (2021) PDF Selected as a 2021 HOT PCCP article
J. Meyer, V. Tajti, E. Carrascosa, T. Győri, M. Stei, T. Michaelsen, B. Bastian, G. Czakó, and R. Wester
Atomistic dynamics of elimination and nucleophilic substitution disentangled for the F− + CH3CH2Cl reaction
Nat. Chem. 13, 977 (2021) PDF Publication of the Month (Chemical Sciences Section, Hungarian Academy of Sciences)
Publication of the Year (University of Szeged)
D. Papp, J. Li, H. Guo, and G. Czakó
Vibrational mode-specificity in the dynamics of the Cl + C2H6 __> HCl + C2H5 reaction
J. Chem. Phys. 155, 114303 (2021) PDF
V. Tajti, T. Győri, and G. Czakó
Detailed quasiclassical dynamics of the F− + CH3Br reaction on an ab initio analytical potential energy surface
J. Chem. Phys. 155, 124301 (2021) PDF
D. Papp and G. Czakó
Vibrational mode-specific dynamics of the F(2P3/2) + C2H6 __> HF + C2H5 reaction
J. Chem. Phys. 155, 154302 (2021) PDF
D. A. Tasi and G. Czakó
Uncovering an oxide ion substitution for the OH− + CH3F reaction
Chem. Sci. 12, 14369 (2021) Cover PDF Selected for the 2021 Chemical Science HOT Article Collection
A. Á. Dékány, G. Z. Kovács, and G. Czakó
High-level systematic ab initio comparison of carbon- and silicon-centered SN2 reactions
J. Phys. Chem. A 125, 9645 (2021) PDF
A. Á. Dékány and G. Czakó
Benchmark ab initio proton affinity and gas-phase basicity of α-alanine based on coupled-cluster theory and statistical mechanics
J. Comput. Chem. 43, 19 (2022) PDF
Z. Kerekes, D. A. Tasi, and G. Czakó
SN2 reactions with an ambident nucleophile: A benchmark ab initio study of the CN− + CH3Y [Y = F, Cl, Br, and I] systems
J. Phys. Chem. A 126, 889 (2022) PDF
T. Győri and G. Czakó
ManyHF: A pragmatic automated method of finding lower-energy Hartree−Fock solutions for potential energy surface development
J. Chem. Phys. 156, 071101 (2022) Communication Cover PDF Selected as Featured
V. Tajti and G. Czakó
Vibrational mode-specific dynamics of the F− + CH3CH2Cl multi-channel reaction
Phys. Chem. Chem. Phys. 24, 8166 (2022) PDF
D. Papp and G. Czakó
Rotational mode-specificity in the Cl + C2H6 __> HCl + C2H5 reaction
J. Phys. Chem. A 126, 2551 (2022) PDF
P. Tóth, T. Szűcs, and G. Czakó
Benchmark ab initio characterization of the abstraction and substitution pathways of the Cl + CH3CN reaction
J. Phys. Chem. A 126, 2802 (2022) PDF
D. A. Tasi and G. Czakó
Unconventional SN2 retention pathways induced by complex formation: High-level dynamics investigation of the NH2− + CH3I polyatomic reaction
J. Chem. Phys. 156, 184306 (2022) PDF
B. Gruber, V. Tajti, and G. Czakó
Full-dimensional automated potential energy surface development and dynamics for the OH + C2H6 reaction
J. Chem. Phys. 157, 074307 (2022) PDF
T. Szűcs and G. Czakó
Benchmark ab initio potential energy surface mapping of the F + CH3NH2 reaction
Phys. Chem. Chem. Phys. 24, 20249 (2022) PDF
C. Yin, V. Tajti, and G. Czakó
Full-dimensional potential energy surface development and dynamics for the HBr + C2H5 __> Br(2P3/2) + C2H6 reaction
Phys. Chem. Chem. Phys. 24, 24784 (2022) PDF Selected as a 2022 HOT PCCP article
C. Yin and G. Czakó
Automated full-dimensional potential energy surface development and quasi-classical dynamics for the HI(X1Σ+) + C2H5 __> I(2P3/2) + C2H6 reaction
Phys. Chem. Chem. Phys. 24, 29084 (2022) PDF
A. B. Nacsa and G. Czakó
Benchmark ab initio determination of the conformers, proton affinities, and gas-phase basicities of cysteine
J. Phys. Chem. A 126, 9667 (2022) PDF
D. Papp, V. Tajti, G. Avila, E. Mátyus, and G. Czakó
CH4.F− revisited: full-dimensional ab initio potential energy surface and variational vibrational states
Mol. Phys. 121, e2113565 (2023) PDF
C. Yin and G. Czakó
Theoretical vibrational mode-specific dynamics studies for the HBr + C2H5 reaction
Phys. Chem. Chem. Phys. 25, 3083 (2023) PDF
D. A. Tasi, T. Michaelsen, R. Wester, and G. Czakó
Quasi-classical trajectory study of the OH− + CH3I reaction: Theory meets experiment
Phys. Chem. Chem. Phys. 25, 4005 (2023) PDF
T. Győri and G. Czakó
A comprehensive benchmark ab initio survey of the stationary points and products of the OH· + CH3OH system
J. Chem. Phys. 158, 034301 (2023) PDF
B. Gruber and G. Czakó
High-level ab initio mapping of the multiple H-abstraction pathways of the OH + glycine reaction
Phys. Chem. Chem. Phys. 25, 5271 (2023) PDF
A. B. Nacsa, M. Kígyósi, and G. Czakó
Protonation of serine: Conformers, proton affinities and gas-phase basicities at the "gold standard" and beyond
Phys. Chem. Chem. Phys. 25, 8891 (2023) PDF
C. Yin and G. Czakó
Vibrational mode-specific quasi-classical trajectory studies for the two-channel HI + C2H5 reaction
Phys. Chem. Chem. Phys. 25, 9944 (2023) PDF
A. B. Nacsa, V. Tajti, and G. Czakó
Dynamics of the Cl− + CH3I reaction on a high-level ab initio analytical potential energy surface
J. Chem. Phys. 158, 194306 (2023) PDF
A. Á. Dékány and G. Czakó
Exploring the versatile reactivity of the F− + SiH3Cl system on a full-dimensional coupled-cluster potential energy surface
J. Chem. Phys. 158, 224303 (2023) PDF
T. Gstir, T. Michaelsen, B. A. Long, A. B. Nacsa, A. Ayasli, D. Swaraj, F. Zappa, F. Trummer, S. G. Ard, N. S. Shuman, G. Czakó, A. A. Viggiano, and R. Wester
The influence of fluorination on the dynamics of the F− + CH3CH2I reaction
Phys. Chem. Chem. Phys. 25, 18711 (2023) PDF Selected as a 2023 HOT PCCP article
C. Yin and G. Czakó
Competition between the H-abstraction and the X-abstraction pathways in the HX (X = Br, I) + C2H5 reactions
Phys. Chem. Chem. Phys. 25, 20241 (2023) PDF Selected as a 2023 HOT PCCP article
A. Giricz, G. Czakó, and D. Papp
Alternating stereospecificity upon central-atom change: Dynamics of the F− + PH2Cl SN2 reaction compared to its C- and N-centered analogues
Chem. Eur. J. 29, e202302113 (2023) PDF Publication of the Month (Chemical Sciences Section, Hungarian Academy of Sciences)
B. Gruber, V. Tajti, and G. Czakó
Vibrational mode-specific dynamics of the OH + C2H6 reaction
J. Phys. Chem. A 127, 7364 (2023) PDF
T. Szűcs and G. Czakó
ManyHF-based full-dimensional potential energy surface development and quasi-classical dynamics for the Cl + CH3NH2 reaction
J. Chem. Phys. 159, 134306 (2023) PDF
C. Yin and G. Czakó
Full-dimensional automated potential energy surface development and detailed dynamics for the CH2OO + NH3 reaction
Phys. Chem. Chem. Phys. 25, 26917 (2023) PDF
B. Ballay, T. Szűcs, D. Papp, and G. Czakó
Phosphorus-centered ion-molecule reactions: benchmark ab initio characterization of the potential energy surfaces of the X− + PH2Y [X, Y = F, Cl, Br, I] systems
Phys. Chem. Chem. Phys. 25, 28925 (2023) PDF
D. A. Tasi and G. Czakó
Vibrational mode-specificity in the dynamics of the OH− + CH3I multi-channel reaction
J. Chem. Phys. 160, 044305 (2024) PDF
T. Szűcs and G. Czakó
Automated potential energy surface development and comprehensive dynamics for the F + CH3NH2 reaction
J. Chem. Phys. 160, 064304 (2024) PDF Editor's Pick
A. B. Nacsa, C. Tokaji, and G. Czakó
High-level analytical potential-energy-surface-based dynamics of the OH− + CH3CH2Cl SN2 and E2 reactions in full (24) dimensions
Faraday Discuss. 251, 604 (2024) PDF
A. Á. Dékány and G. Czakó
Detailed quasiclassical dynamics of the F− + SiH3Cl multi-channel reaction
Phys. Chem. Chem. Phys. 26, 10008 (2024) PDF
A. Ayasli, P. Tóth, T. Michaelsen, T. Gstir, F. Zappa, D. Papp, G. Czakó, and R. Wester
Imaging the ion-molecule reaction dynamics of O− + CD4
J. Phys. Chem. A 128, 3078 (2024) PDF
G. Czakó, B. Gruber, D. Papp, V. Tajti, D. A. Tasi, and C. Yin
First-principles mode-specific reaction dynamics
Phys. Chem. Chem. Phys. 26, 15818 (2024) Perspective (PCCP 25th Anniversary Collection) Cover PDF Selected as a 2024 HOT PCCP article
D. A. Tasi and G. Czakó
Benchmark ab initio characterization of the complex potential energy surfaces of the HOO− + CH3Y [Y = F, Cl, Br, I] reactions
Phys. Chem. Chem. Phys. 26, 16048 (2024) PDF
K. Horváth, V. Tajti, D. Papp, and G. Czakó
Dynamics of the HCl + C2H5 multi-channel reaction on a full-dimensional ab initio potential energy surface
J. Phys. Chem. A 128, 4474 (2024) PDF
D. R. Gál, D. Papp, and G. Czakó
Benchmark ab initio characterization of the multi-channel Cl + CH3X [X = F, Cl, Br, I] reactive potential energy surfaces
Phys. Chem. Chem. Phys. 26, 17695 (2024) PDF
C. Yin and G. Czakó
Revealing new pathways for the reaction of Criegee intermediate CH2OO with SO2
Commun. Chem. 7, 157 (2024) PDF
P. Tóth, T. Szűcs, T. Győri, and G. Czakó
Dynamics of the Cl + CH3CN reaction on an automatically-developed full-dimensional ab initio potential energy surface
J. Chem. Phys. 161, 084304 (2024) PDF
E. Mátyus and G. Czakó
Foreword to the Festschrift in honour of Professor Attila G. Császár: molecules (always) in motion
Mol. Phys. 122, e2360850 (2024) PDF
F. J. Aoiz, N. Balucani, A. Bergeat, A. Butler, D. W. Chandler, G. Czakó, T. Győri, D. E. Heard, D. Heathcote, B. R. Heazlewood, N. Hertl, P. G. Jambrina, R. I. Kaiser, O. A. Krohn, V. Le Duc, J. Loreau, S. R. Mackenzie, K. G. McKendrick, J. Meyer, G. M. Nathanson, D. M. Neumark, R. Pandey, C. Reilly, P. Robertson, G. C. Schatz, S. J. Sibener, A. G. Suits, P. D. Watson, R. Wester, S. Willitsch, A. M. Wodtke, and B. S. Zhao
Scattering of larger molecules – part 2: general discussion
Faraday Discuss. 251, 622 (2024) PDF
B. Gruber and G. Czakó
High-level ab initio characterization of the OH + CH3NH2 reaction
Phys. Chem. Chem. Phys. 26, 28543 (2024) PDF
B. J. Molnár, A. Á. Dékány, and G. Czakó
Automated potential energy surface development and quasi-classical dynamics for the F− + SiH3I system
J. Chem. Phys. 161, 194306 (2024) PDF
D. A. Tasi, E. M. Orján, and G. Czakó
Benchmark ab initio mapping of the F− + CH2ClI SN2 and proton-abstraction reactions
J. Phys. Chem. A 128, 10568 (2024) PDF
G. Czakó
First-principles dynamics and mechanisms of fundamental chemical reactions
Adv. Quantum Chem. in press (2025) Book chapter
A. Ayasli, A. Khan, T. Gstir, T. Michaelsen, D. Papp, Y. Wang, H. Song, M. Yang, G. Czakó, and R. Wester
A dynamic isotope effect in the nucleophilic substitution reaction between F− and CD3I
Nat. Commun. 16, 2318 (2025) PDF
Y. Wang, Z. Tu, G. Czakó, H. Song, and M. Yang
Mode-specific quantum and quasi-classical trajectory dynamics of the F− + CH3I __> I− + CH3F SN2 reaction
J. Phys. Chem. A DOI: 10.1021/acs.jpca.5c03163 (2025)
Presentations
Lectures
A. G. Császár, V. Szalay, and G. Czakó
First-principles rovibrational spectroscopy
Conference on the Interfaces among Mathematics, Chemistry & Computer Sciences, Dubrovnik, Croatia, June 2004
G. Czakó, A. G. Császár, and V. Szalay
Toward first-principles complete spectroscopy of small molecules
The 2005 Younger European Chemists’ Conference, Brno, Czech Republic, September 2005
G. Czakó, V. Szalay, and A. G. Császár
Szingularitások kezelése háromtest problémák variációs megoldásai során (in Hungarian)
XI. MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, May 2006
T. Furtenbacher, G. Czakó, A. G. Császár, and V. Szalay
Kismolekulák teljes spektroszkópiája (in Hungarian)
XI. MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, May 2006
E. Mátyus, G. Czakó, T. Furtenbacher, J. Simunek, and A. G. Császár
High-accuracy high-resolution variationally computed molecular spectra
22nd Austin Symposium on Molecular Structure, Austin, TX, USA, March 2008
G. Czakó, E. Mátyus, A. G. Császár, B. J. Braams, and J. M. Bowman
High-accuracy ab initio rovibrational spectroscopy
Southeastern Theoretical Chemistry Association (SETCA), University of Alabama, Tuscaloosa, AL, USA, May 2008
A. G. Császár, T. Furtenbacher, E. Mátyus, G. Czakó, C. Fábri, and T. Szidarovszky
Spectropedia
Molecular Modeling in Chemistry and Biochemistry, Kolozsvár, Romania, April 2009
E. Mátyus, G. Czakó, and A. G. Császár
Általánosított módszerek variációs alapú magmozgás számításokhoz (in Hungarian)
XII. MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, May 2009
C. Fábri, G. Czakó, and A. G. Császár
Túl a Born-Oppenheimer közelítésen: A H2+ izotopológjainak rezgései (in Hungarian)
XII. MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, May 2009
T. Szidarovszky, G. Czakó, and A. G. Császár
Effektív algoritmus 3-atomos rendszerek rezgési-forgási színképének számítására (in Hungarian)
XII. MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, May 2009
G. Czakó, B. C. Shepler, B. J. Braams, and J. M. Bowman
Accurate ab initio potential energy surface, dynamics, and thermochemistry of the F + CH4 __> HF + CH3 reaction
Southeastern Theoretical Chemistry Association (SETCA), Duke University, Durham, NC, USA, May 2009
G. Czakó, B. C. Shepler, B. J. Braams, and J. M. Bowman
Accurate ab initio potential energy surface, dynamics, and thermochemistry of the F + CH4 __> HF + CH3 reaction
XXII Dynamics of Molecular Collisions Meeting, Snowbird, UT, USA, July 2009
J. M. Bowman, C. Chen, B. C. Shepler, G. Czakó, and B. J. Braams
Reaction dynamics using full dimensional ab initio potential energy surfaces
238th ACS National Meeting, Washington, DC, USA, August 16-20 2009
G. Czakó, B. C. Shepler, B. J. Braams, A. L. Kaledin, and J. M. Bowman
Quasiclassical dynamics of the F + CHD3 reaction and the water dimer
Southeastern Theoretical Chemistry Association (SETCA), University of South Carolina, Columbia, SC, USA, May 2010
J. M. Bowman, G. Czakó, B. Fu, and Y. Han
Reaction dynamics calculations on high dimensional, ab initio potential energy surfaces
XXIII Dynamics of Molecular Collisions Meeting, Snowbird, UT, USA, July 2011
J. M. Bowman, G. Czakó, and B. Fu
Reaction dynamics on high-dimensional ab initio potential energy surfaces
XI International Workshop on Quantum Reactive Scattering, Santa Fe, NM, USA, July 2011
G. Czakó
Dynamics of fundamental bimolecular chemical reactions (in Hungarian)
MTA Material and Molecular Structure Working Group Meeting, Szedres, Hungary, October 12-14 2012
G. Czakó
Dynamics of six-atom reactions on chemically accurate ab initio potential energy surfaces
XII International Workshop on Quantum Reactive Scattering, Bordeaux, France, June 10-14 2013 (Invited talk)
G. Czakó
Dynamics of bimolecular polyatomic reactions on ab initio potential energy surfaces
XXIV Dynamics of Molecular Collisions Meeting, Granlibakken, CA, USA, July 7-12 2013
G. Czakó
Rotational-vibrational and reaction dynamics of polyatomic systems on ab initio potential energy surfaces
Kick-Off Meeting of Working Group 5 of the COST CoDECS Action, Budapest, Hungary, August 30-31 2013 (Invited talk)
G. Czakó
Hatatomos rendszerek dinamikája (in Hungarian)
MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, October 17-19 2013
G. Czakó
Kémiai reakciók dinamikája ab initio potenciális energia felületeken (in Hungarian)
MTA Physical Chemistry Scientific Committee Meeting, Budapest, Hungary, October 24 2013
P. Szabó, G. Czakó, and G. Lendvay
Dynamics of the reaction HBr + CH3 __> Br + CH4 on a global ab initio potential energy surface: the origin of the negative activation energy
23rd International Symposium on Gas Kinetics and Related Phenomena, Szeged, Hungary, July 20-25 2014
G. Czakó
Dynamics of polyatomic chemical reactions on ab initio potential energy surfaces
20th European Conference on the Dynamics of Molecular Systems (MOLEC), Gothenburg, Sweden, August 24-29 2014
G. Czakó
Dynamics of polyatomic chemical reactions on ab initio analytical potential energy surfaces
Institutional seminar, Institute for Ion Physics and Applied Physics, University of Innsbruck, Innsbruck, Austria, November 12 2014 (Invited talk)
G. Czakó
Kémiai reakciók dinamikája ab initio potenciális energia felületeken (in Hungarian)
MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, February 27-28 2015
G. Czakó
Dynamics of hydrogen-abstraction and SN2 reactions on ab initio analytical potential energy surfaces
XIII International Workshop on Quantum Reactive Scattering, Salamanca, Spain, July 6-10 2015 (Invited talk)
P. Szabó, G. Lendvay, and G. Czakó
Quasiclassical trajectory studies of the dynamics of a complex-forming bimolecular reaction: CH3 + HBr __> CH4 + Br
XIII International Workshop on Quantum Reactive Scattering, Salamanca, Spain, July 6-10 2015
M. Stei, E. Carrascosa, M. Kainz, A. Kelkar, I. Szabó, G. Czakó, A. Dörfler, T. Michaelsen, J. Meyer, and R. Wester
Imaging the influence of the leaving group and of vibrational excitation on SN2 reactions
XXV Dynamics of Molecular Collisions Meeting, Asilomar, CA, USA, July 12-17 2015
G. Czakó
Dynamics of chemical reactions on ab initio potential energy surfaces
14th Central European Symposium on Theoretical Chemistry (CESTC), Banská Bystrica, Slovakia, September 6-9 2015 (Invited talk)
G. Czakó
Kémiai reakciók dinamikája ab initio potenciális energia felületeken (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Balatonalmádi, Hungary, May 29-30 2017
G. Czakó
Dynamics and novel mechanisms of SN2 reactions on ab initio analytical potential energy surfaces
8th International Meeting on Atomic and Molecular Physics and Chemistry (IMAMPC), Torun, Poland, June 19-22 2017 (Invited talk)
G. Czakó
Dynamics and novel mechanisms of SN2 reactions on ab initio analytical potential energy surfaces
XIV International Workshop on Quantum Reactive Scattering, Trieste, Italy, July 3-6 2017 (Invited talk)
G. Czakó
Dynamics and novel mechanisms of SN2 reactions on ab initio analytical potential energy surfaces
SN2 Reaction Dynamics Symposium, ACS Southwest Regional Meeting, Lubbock, TX, USA, October 29 - November 1 2017 (Invited talk)
G. Czakó
Dynamics and novel mechanisms of reactions of atoms and ions with polyatomic molecules
WG1 Workshop, COST Action CM1401, Ciudad Real, Spain, December 11-12 2017 (Invited talk)
G. Czakó
Dynamics and novel mechanisms of reactions of atoms and ions with polyatomic molecules
3rd Anharmonicity in medium-sized molecules and clusters (AMOC) Meeting, Budapest, Hungary, April 16-19 2018 (Invited talk)
D. Papp, G. Balázs, and G. Czakó
Halogénatom + etán reakciók potenciálisenergia-felületeinek nagypontosságú ab initio feltérképezése (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Veszprém, Hungary, November 8-9 2018
D. A. Tasi, Z. Fábián, and G. Czakó
Az X− + CH3Y [X = OH, SH, CN, PH2, NH2; Y = F, Cl, Br, I] SN2 reakciók inverziós és retenciós reakcióútjainak nagypontosságú ab initio feltérképezése (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Veszprém, Hungary, November 8-9 2018
V. Tajti and G. Czakó
A F− + CH3CH2Cl reakció komplex potenciálisenergia-felületének nagypontosságú ab initio feltérképezése (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Veszprém, Hungary, November 8-9 2018
T. Győri and G. Czakó
Potenciálisenergia-felületek fejlesztésének automatizálása (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Veszprém, Hungary, November 8-9 2018
B. Olasz and G. Czakó
A F− + CH3I reakció dinamikájának és mikroszolvatációjának elméleti vizsgálata (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Veszprém, Hungary, November 8-9 2018
D. A. Tasi and G. Czakó
Különböző nukleofilek metil halogenidekkel történő SN2 reakcióinak elméleti vizsgálata (in Hungarian)
I. FKF symposium, Debrecen, Hungary, April 3-5 2019
G. Czakó
Elméleti reakciódinamika kutatások a Szegedi Tudományegyetemen (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, June 6-7 2019
G. Czakó
Dynamics and mechanisms of complex chemical reactions on ab initio analytical potential energy surfaces
XV International Workshop on Quantum Reactive Scattering, Tokyo (Saitama), Japan, July 1-5 2019 (Invited talk)
D. A. Tasi and G. Czakó
A OH− + CH3I reakció potenciálisenergia-felületének fejlesztése különböző kvantumkémiai szinteken (in Hungarian)
Chemistry Lecture Days, Szeged, Hungary, October 28-30 2019
D. A. Tasi and G. Czakó
A OH− + CH3I reakció potenciálisenergia-felületének fejlesztése és dinamikája különböző kvantumkémiai szinteken (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Mátrafüred, Hungary, November 7-8 2019
G. Czakó
Elméleti reakciódinamika kutatások a Szegedi Tudományegyetemen (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Mátrafüred, Hungary, November 7-8 2019
V. Tajti and G. Czakó
Ion-molekula reakciók mechanizmusainak tanulmányozása (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Online, November 6 2020
G. Czakó
Reaction dynamics on analytical potential energy surfaces
"Dynamics of Chemical Reactions" Lecture Series of the Beijing Institute of Technology, Online, January 4 2021 (Invited talk)
G. Czakó
First-principles reaction dynamics beyond six-atom systems
Dynamics of Chemical Reactions from Gas Phase to Interfaces, A Symposium in Honor of Professor William L. Hase, 2021 Spring ACS Meeting, Online, April 12-15 2021 (Invited talk)
T. Győri and G. Czakó
Ab initio számítások a metanol + hidroxilgyök rendszerre és praktikák a Hartree-Fock konvergenciaproblémák ellen (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Online, May 27 2021
D. A. Tasi and G. Czakó
Poliatom-poliatom típusú SN2 reakció dinamikájának vizsgálata: az NH2− + CH3I reakció esete (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Online, May 27 2021
A. Dékány and G. Czakó
Az α-alanin protonaffinitásának és gázfázisú bázicitásának nagy pontosságú elméleti kémiai meghatározása (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Online, May 27 2021
G. Czakó
Automated potential energy surface developments for chemical reaction dynamics simulations
Machine Learning and Informatics for Chemistry and Materials, Telluride Workshop, Online, September 27 - October 1 2021 (Invited talk)
D. Papp and G. Czakó
Atom + etán és nitrogén-centrumú SN2 reakciók dinamikája (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, September 30 - October 1 2021
V. Tajti and G. Czakó
Számítógéppel az ion-molekula reakciók mechanizmusainak nyomában (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, September 30 - October 1 2021
A. B. Nacsa and G. Czakó
A glicin protonaffinitásának nagypontosságú ab initio meghatározása (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, September 30 - October 1 2021
G. Czakó
Newton almájától a kémiai reakciók dinamikájáig (in Hungarian)
Institutional seminar, Institute of Physics, University of Pécs, Pécs, Hungary, October 6 2021 (Invited talk)
D. Papp and G. Czakó
Atom + etán és nitrogén-centrumú SN2 reakciók dinamikája (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Mátrafüred, Hungary, October 21-22 2021
B. Gruber and G. Czakó
Az OH gyök reakciója metán, etán és glicin molekulákkal (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Mátrafüred, Hungary, October 21-22 2021
D. A. Tasi and G. Czakó
A OH− + CH3F reakció dinamikai vizsgálata: az oxidion szubsztitúció (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Mátrafüred (online), Hungary, October 21-22 2021
T. Szűcs and G. Czakó
A Cl + CH3NH2 reakció potenciálisenergia-felületének nagypontosságú ab initio feltérképezése (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Mátrafüred, Hungary, October 21-22 2021
V. Tajti, T. Győri, and G. Czakó
Az elmélet és kísérlet együttműködése: a F− + CH3CH2Cl szubsztitúciós és eliminációs reakciók versengő dinamikája (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Balatonvilágos (online), Hungary, May 19-20 2022
A. Giricz, D. Papp, and G. Czakó
A foszfor, mint központi atom SN2 és proton-transzfer reakcióban: a F− + PH2Cl reakció dinamikájának elméleti modellezése (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Balatonvilágos (online), Hungary, May 19-20 2022
P. Tóth, T. Szűcs, and G. Czakó
A Cl + CH3CN reakció absztrakciós és szubsztitúciós útvonalainak nagypontosságú kvantumkémiai feltérképezése (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Balatonvilágos (online), Hungary, May 19-20 2022
A. Á. Dékány and G. Czakó
SN2 reakciók szilícium centrumon: a F− + SiH3Cl rendszer dinamikája (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, June 2-3 2022
D. A. Tasi and G. Czakó
Rendhagyó reakcióút OH− + CH3F esetén: az oxidion szubsztitúció (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, June 2-3 2022
T. Győri and G. Czakó
Jobb Hartree−Fock megoldások keresésének automatizálása a ManyHF módszerrel (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, June 2-3 2022
T. Győri, D. A. Tasi, V. Tajti, D. Papp, and G. Czakó
Towards automated potential energy surface development with Robosurfer and ManyHF
Gordon Research Conference on Molecular Interactions and Dynamics, Stonehill College, Easton, MA, USA, July 10-15 2022
T. Győri, D. A. Tasi, V. Tajti, D. Papp, and G. Czakó
Tools for automated PES development: Robosurfer and ManyHF
XVI International Workshop on Quantum Reactive Scattering, Balatonföldvár, Hungary, September 4-9 2022
D. A. Tasi and G. Czakó
Dynamics of the OH− + CH3F reaction: The oxide ion substitution
XVI International Workshop on Quantum Reactive Scattering, Balatonföldvár, Hungary, September 4-9 2022
G. Czakó
Computational reaction dynamics at the University of Szeged
MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, October 21-22 2022
A. B. Nacsa, E. M. Orján, and G. Czakó
Ab initio conformational analysis of dehydrogenated and protonated amino acids
MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, October 21-22 2022
T. Győri, D. A. Tasi, V. Tajti, D. Papp, and G. Czakó
Towards automated potential energy surface development with Robosurfer and ManyHF
MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, October 21-22 2022
D. Papp, V. Tajti, G. Avila, E. Mátyus, and G. Czakó
A CH4.Ar és a CH4.F− komplexek rezgési dinamikája spektroszkópiai minőségű potenciálisenergia-felületeken (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Mátrafüred, Hungary, October 27-28 2022
A. B. Nacsa, E. M. Orján, and G. Czakó
Dehidrogénezett és protonált aminosavak ab initio konformeranalízise (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Mátrafüred, Hungary, October 27-28 2022
B. Gruber, V. Tajti, and G. Czakó
Az OH + C2H6 reakció dinamikája egy automatikusan fejlesztett potenciálisenergia-felületen (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Mátrafüred, Hungary, October 27-28 2022
D. Papp, A. Giricz, and G. Czakó
Vibrational-, rotational- and stereo-specificity in the dynamics of atom + ethane and N/P centered SN2 reactions
Stereodynamics Conference, Rethymnon, Crete, Greece, October 30 - November 4 2022 (Invited talk of D. Papp)
G. Czakó
Kémiai reakciók kísérleti ujjlenyomatainak elméleti megfejtése (in Hungarian)
Lecture day of the Chemistry Commission celebrating Hungarian Science, MTA-SZAB Hall, Szeged, Hungary, November 16 2022 (Invited talk)
P. Tóth, T. Szűcs, and G. Czakó
A Cl + CH3CN reakció absztrakciós és szubsztitúciós útvonalainak nagypontosságú kvantumkémiai feltérképezése (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, May 18-19 2023
B. I. Sipos, B. Gruber, and G. Czakó
Az O(3P) + CH4 reakció dinamikájának vizsgálata egy új ab initio globális potenciálisenergia-felületen (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, May 18-19 2023
C. Yin and G. Czakó
Automated full-dimensional potential energy surface development and quasi-classical dynamics for the HX + C2H5 (X = Br, I) reactions
KeMoMo-QSAR symposium, Szeged, Hungary, May 18-19 2023
T. Szűcs and G. Czakó
A Cl + CH3NH2 reakció dinamikájának elméleti modellezése (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Balatonvilágos, Hungary, June 8-9 2023
A. Á. Dékány and G. Czakó
A F− + SiH3Cl reakció dinamikája analitikus potenciálisenergia-felületen (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Balatonvilágos, Hungary, June 8-9 2023
P. Tóth, T. Szűcs, and G. Czakó
A Cl + CH3CN reakció mechanizmusainak és dinamikájának elméleti modellezése (in Hungarian)
Chemistry Lecture Days, Szeged, Hungary, October 17-19 2023
G. Czakó
First-principles dynamics and mechanisms of gas-phase bimolecular chemical reactions
International Conference on Molecular Energy Transfer in Complex Systems (iCOMET), Shiv Vilas Resorts, Jaipur, India, November 12-17 2023 (Invited talk)
D. A. Tasi and G. Czakó
A OH− + CH3F/CH3I reakciók dinamikájának elméleti vizsgálta (in Hungarian)
MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, November 24-25 2023
A. B. Nacsa, C. Tokaji, and G. Czakó
Dynamics of competing SN2 and E2 reactions
MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, November 24-25 2023
T. Szűcs and G. Czakó
Automated potential energy surface developments and dynamics simulations for the Cl/F + CH3NH2 reactions
MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, November 24-25 2023
B. Gruber and G. Czakó
Csatolt-klaszter minőségű potenciálisenergia-felület fejlesztése és a dinamika tanulmányozása teljes (30) dimenzióban: a glicin + OH reakció (in Hungarian)
MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, November 24-25 2023
G. Czakó
First-principles dynamics and mechanisms of fundamental organic reactions
"Highlighting Organic Chemistry in Hungary" Webinar, European Chemical Society, Division of Organic Chemistry, March 1 2024 (Invited talk)
A. B. Nacsa, C. Tokaji, and G. Czakó
High-level analytical potential-energy-surface-based dynamics of the OH− + CH3CH2Cl SN2 and E2 reactions in full (24) dimensions
New directions in molecular scattering Faraday Discussion, Edinburgh, United Kingdom (Online), May 8-10 2024 (Invited talk)
D. Papp, L. Erdei, D. Gál, A. Giricz, R. Gyimesi, and G. Czakó
A központi atom hatásának elméleti vizsgálata ion–molekula reakciók dinamikájában (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Balatonvilágos, Hungary, May 30-31 2024
T. Szűcs and G. Czakó
SN2 reakció két reaktív centrummal: A CN− +CH3I rendszer dinamikája egy globális potenciálisenergia-felületen (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Balatonvilágos, Hungary, May 30-31 2024
D. A. Tasi, E. Orján, and G. Czakó
Bimolekuláris nukleofil szubsztitúció két lehetséges távozócsoporttal: a F− + CH2ClI reakció elméleti vizsgálata (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, June 3-4 2024
D. Papp, L. Erdei, D. Gál, A. Giricz, R. Gyimesi, and G. Czakó
A központi atom és a távozócsoport hatásának elméleti modellezése SN2 reakciók dinamikájában (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, June 3-4 2024
G. Czakó
First-principles reaction dynamics from 5 to 12 atoms
XVII International Workshop on Quantum Reactive Scattering, Istanbul, Türkiye, June 24-28 2024 (Invited talk)
G. Czakó
Automated ab initio potential energy surface developments for reaction dynamics computations
International Conference on Molecular Electronic Structure, Pescara, Italy, September 21-25 2024 (Invited talk)
P. Tóth, T. Szűcs, and G. Czakó
A Cl + CH3CN reakció dinamikájának vizsgálata automatikusan fejlesztett teljes dimenziós ab initio potenciálisenergia-felületen (in Hungarian)
Chemistry Lecture Days, Szeged, Hungary, October 29-31 2024 (Invited talk of P. Tóth)
R. S. Gyimesi, D. Papp, and G. Czakó
A F− + NH2I reakció analitikus potenciálisenergia-felületének kifejlesztése és mechanizmusainak részletes vizsgálata (in Hungarian)
Chemistry Lecture Days, Szeged, Hungary, October 29-31 2024
D. R. Gál, D. Papp, and G. Czakó
A Cl + CH3X (X= F, Cl, Br, I) reakciók lehetséges útvonalainak nagypontosságú kvantumkémiai feltérképezése (in Hungarian)
Chemistry Lecture Days, Szeged, Hungary, October 29-31 2024
L. C. Erdei, D. Papp, and G. Czakó
A F− + PH2I reakció dinamikájának modellezése saját fejlesztésű ab initio potenciálisenergia-felületen (in Hungarian)
Chemistry Lecture Days, Szeged, Hungary, October 29-31 2024
B. Imre, B. Gruber, and G. Czakó
Etanol és klóratom reakciójának ab initio feltérképezése (in Hungarian)
Chemistry Lecture Days, Szeged, Hungary, October 29-31 2024
P. Tóth, T. Szűcs, and G. Czakó
A Cl + CH3CN reakció dinamikájának vizsgálata automatikusan fejlesztett teljes dimenziós ab initio potenciálisenergia-felületen (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Mátrafüred, Hungary, November 7-8 2024
D. R. Gál, D. Papp, and G. Czakó
Cl + CH3X (X = F, Cl, Br, I) reakcióutak kvantumkémiai feltérképezése és a Cl + CH3F rendszer analitikus potenciálisenergia-felületének fejlesztése (in Hungarian)
MTA Reaction Kinetics and Photochemistry Working Group Meeting, Mátrafüred, Hungary, November 7-8 2024
G. Czakó
From stationary-point properties to reaction dynamics: A special overview
MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, November 22-23 2024
A. Sunaga, G. Avila, E. Mátyus, T. Győri, V. Tajti, and G. Czakó
Variational vibrational states of methanol and the development of a new potential energy surface
MTA Material and Molecular Structure Working Group Meeting, Mátrafüred, Hungary, November 22-23 2024
T. Győri and G. Czakó
Spektroszkópiai pontosságú potenciálfelület fejlesztése a metanol teljes dimenziós rezgési modellezéséhez (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, May 29-30 2025
P. Tóth and G. Czakó
Reakciódinamika és izotóphatás az O− + CH4/CD4 rendszerekben: mélyebb betekintés a kísérleti megfigyelésekbe elméleti módszerekkel (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, May 29-30 2025
B. J. Molnár, A. Á. Dékány, and G. Czakó
A F− + SiH3I reakció dinamikájának elméleti modellezése (in Hungarian)
KeMoMo-QSAR symposium, Szeged, Hungary, May 29-30 2025
Posters
G. Czakó, A. G. Császár, and V. Szalay
Toward first-principles complete spectroscopy of small molecules
The 2005 Younger European Chemists’ Conference, Brno, Czech Republic, September 2005
E. Mátyus, G. Czakó, B. T. Sutcliffe, and A. G. Császár
Variational vibrational calculations in normal coordinates with arbitrary potentials
Molecular Quantum Mechanics: Analytic Gradients and Beyond, An International Conference in Honor of Professor Péter Pulay, Budapest, Hungary, May 2007
G. Czakó, T. Furtenbacher, A. G. Császár, and V. Szalay
Toward first-principles complete spectroscopy of small molecules
Molecular Quantum Mechanics: Analytic Gradients and Beyond, An International Conference in Honor of Professor Péter Pulay, Budapest, Hungary, May 2007
G. Czakó, C. Fábri, A. G. Császár, V. Szalay, B. T. Sutcliffe, and G. Tasi
Adiabatic Jacobi corrections for H2+-like systems
Molecular Quantum Mechanics: Analytic Gradients and Beyond, An International Conference in Honor of Professor Péter Pulay, Budapest, Hungary, May 2007
G. Czakó, C. Fábri, T. Szidarovszky, and A. G. Császár
Variational spectroscopy of H2+ and H3+ and their isotopologues
NATO ARW Hydrogen: A universal saga, Brijuni Island, Croatia, August 2008
E. Mátyus, G. Czakó, J. Simunek, and A. G. Császár
Universal approaches to the variational nuclear motion problem
20th International Conference on High-Resolution Molecular Spectroscopy, Praha, Czech Republic, September 2008
G. Czakó, B. C. Shepler, B. J. Braams, and J. M. Bowman
Accurate ab initio potential energy surface, dynamics, and thermochemistry of the F + CH4 __> HF + CH3 reaction
Gordon Research Conference on Molecular Energy Transfer, Ventura, CA, USA, January 2009
G. Czakó and J. M. Bowman
State-to-state quasiclassical dynamics of the F + CH4 and F + CHD3 reactions
International Chemical Congress of Pacific Basin Societies (PACIFICHEM), Honolulu, Hawaii, USA, December 2010
G. Czakó and J. M. Bowman
Accurate ab initio potential energy surfaces and dynamics for the reactions of methane with F and Cl atoms
XXIII Dynamics of Molecular Collisions Meeting, Snowbird, UT, USA, July 2011
G. Czakó and J. M. Bowman
Accurate ab initio potential energy surfaces and dynamics for the reactions of methane with F and Cl atoms
XI International Workshop on Quantum Reactive Scattering, Santa Fe, NM, USA, July 2011
G. Czakó and J. M. Bowman
Reaction dynamics on accurate ab initio potential energy surfaces
Anharmonicity in medium-sized molecules and clusters, Université Paris-Est Marne-La-Vallée, France, April 18-21 2012
T. Szidarovszky, A. G. Császár, and G. Czakó
Rovibrational spectra near dissociation
Anharmonicity in medium-sized molecules and clusters, Université Paris-Est Marne-La-Vallée, France, April 18-21 2012
G. Czakó, B. J. Braams, J. M. Bowman, C. Fábri, and A. G. Császár
Potential energy surface and rotational-vibrational dynamics of the F−_CH4 anion complex
The 23rd Colloquium on High Resolution Molecular Spectroscopy, Budapest, Hungary, August 25-30 2013
G. Czakó
Dynamics of polyatomic chemical reactions on ab initio potential energy surfaces
23rd International Symposium on Gas Kinetics and Related Phenomena, Szeged, Hungary, July 20-25 2014
G. Czakó
Dynamics of polyatomic chemical reactions on ab initio potential energy surfaces
20th European Conference on the Dynamics of Molecular Systems (MOLEC), Gothenburg, Sweden, August 24-29 2014
G. Czakó
Dynamics of hydrogen-abstraction and SN2 reactions on ab initio analytical potential energy surfaces
XXV Dynamics of Molecular Collisions Meeting, Asilomar, CA, USA, July 12-17 2015
M. Stei, E. Carrascosa, J. Meyer, T. Michaelsen, B. Bastian, M. A. Kainz, A. H. Kelkar, I. Szabó, G. Czakó, and R. Wester
Imaging the complex influence of the leaving group on nucleophilic substitution reactions
12th European Conference on Atoms Molecules and Photons (ECAMP12), Frankfurt, Germany, September 5-9 2016
M. Stei, E. Carrascosa, J. Meyer, T. Michaelsen, B. Bastian, M. A. Kainz, A. H. Kelkar, I. Szabó, G. Czakó, and R. Wester
Imaging the complex influence of the leaving group on nucleophilic substitution reactions
21st European Conference on the Dynamics of Molecular Systems (MOLEC), Toledo, Spain, September 11-16 2016
G. Czakó
Dynamics of bimolecular chemical reactions on ab initio potential energy surfaces
XXVIII International Symposium on Molecular Beams, Edinburgh, Scotland, June 23-28 2019
D. Papp and G. Czakó
Benchmark ab initio study of the X + C2H6 [X = F, Cl, Br, I] systems, dynamics of the X + C2H6 [X = F, Cl] reactions, and PESs for spectroscopy
XXVIII International Symposium on Molecular Beams, Edinburgh, Scotland, June 23-28 2019
D. A. Tasi and G. Czakó
Rethinking fundamental SN2 reactions
XXVIII International Symposium on Molecular Beams, Edinburgh, Scotland, June 23-28 2019
T. Győri and G. Czakó
Automated potential energy surface development: Application to the F− + CH3Br system
XXVIII International Symposium on Molecular Beams, Edinburgh, Scotland, June 23-28 2019
V. Tajti and G. Czakó
Accurate ab initio thermochemistry, potential energy surface, and dynamics of the F− + CH3CH2Cl reaction
XXVIII International Symposium on Molecular Beams, Edinburgh, Scotland, June 23-28 2019
D. Papp and G. Czakó
Benchmark ab initio study of the X + C2H6 [X = F, Cl, Br, I] systems, dynamics of the X + C2H6 [X = F, Cl] reactions, and PESs for spectroscopy
XV International Workshop on Quantum Reactive Scattering, Tokyo (Saitama), Japan, July 1-5 2019
T. Győri, D. A. Tasi, V. Tajti, D. Papp, and G. Czakó
Towards automated potential energy surface development with Robosurfer and ManyHF
Gordon Research Conference on Molecular Interactions and Dynamics, Stonehill College, Easton, MA, USA, July 10-15 2022
A. B. Nacsa, V. Tajti, and G. Czakó
Toward the first-principles dynamics of the Cl− + CH3I and F− + CF3CH2I reactions
Gordon Research Conference on Molecular Interactions and Dynamics, Stonehill College, Easton, MA, USA, July 10-15 2022
D. Papp, A. Giricz, and G. Czakó
Dynamics of atom + ethane and N/P-centered ion-molecule reactions
Gordon Research Conference on Molecular Interactions and Dynamics, Stonehill College, Easton, MA, USA, July 10-15 2022
T. Szűcs, P. Tóth, and G. Czakó
High-level ab initio stationary-point characterizations of atom + molecule multi-channel reactions
Gordon Research Conference on Molecular Interactions and Dynamics, Stonehill College, Easton, MA, USA, July 10-15 2022
V. Tajti, T. Győri, P. Papp, and G. Czakó
Theoretical investigation and numerical separation of competitive reaction pathways for ion-molecule reactions
Gordon Research Conference on Molecular Interactions and Dynamics, Stonehill College, Easton, MA, USA, July 10-15 2022
T. Győri and G. Czakó
Automated development of PESs via active learning: a real-world stress test of electronic structure theories and implementations?
17th International Congress of Quantum Chemistry (ICQC), Bratislava, Slovakia, June 26 – July 1 2023
A. B. Nacsa, V. Tajti, and G. Czakó
Theoretical insights into the Cl− + CH3I and the F− + CF3CH2I reactions
XXVIII Dynamics of Molecular Collisions Conference, Snowbird, UT, USA, July 9-14 2023
B. Gruber, V. Tajti, and G. Czakó
Theoretical investigation of the reactions of OH with ethane and glycine
XXVIII Dynamics of Molecular Collisions Conference, Snowbird, UT, USA, July 9-14 2023
T. Szűcs and G. Czakó
Spin–orbit-corrected full-dimensional ab initio potential energy surface development and dynamics for the Cl/F + CH3NH2 reactions
International Conference on Molecular Energy Transfer in Complex Systems (iCOMET), Shiv Vilas Resorts, Jaipur, India, November 12-17 2023
P. Tóth, T. Szűcs, and G. Czakó
Benchmark ab initio characterization, potential energy surface development and dynamics for the abstraction and substitution pathways of the Cl + CH3CN reaction
International Conference on Molecular Energy Transfer in Complex Systems (iCOMET), Shiv Vilas Resorts, Jaipur, India, November 12-17 2023
B. Gruber and G. Czakó
Exploring the dynamics of the OH + glycine H-abstraction reaction using an in-house-developed analytical potential energy surface
New directions in molecular scattering Faraday Discussion, Edinburgh, United Kingdom, May 8-10 2024
T. Győri and G. Czakó
Automated PES development with Robosurfer and ManyHF: active learning demands robust electronic structure theory and implementation
New directions in molecular scattering Faraday Discussion, Edinburgh, United Kingdom, May 8-10 2024
D. Papp and G. Czakó
Modelling the dynamics of abstraction and substitution processes in polyatomic reactions
XVII International Workshop on Quantum Reactive Scattering, Istanbul, Türkiye, June 24-28 2024
V. Tajti and G. Czakó
Theoretical characterization of the mechanisms of ion-molecule reactions
XVII International Workshop on Quantum Reactive Scattering, Istanbul, Türkiye, June 24-28 2024
P. Tóth, T. Szűcs, and G. Czakó
Benchmark ab initio characterization, automated potential energy surface development and quasi-classical dynamics for the Cl + CH3CN reaction
XVII International Workshop on Quantum Reactive Scattering, Istanbul, Türkiye, June 24-28 2024
P. Tóth, T. Szűcs, and G. Czakó
Ab initio characterization, PES development, and mode-specific quasi-classical dynamics for the Cl + CH3CN and O− + CH4/CD4 reactions
XXIX Dynamics of Molecular Collisions Conference, Snowbird, UT, USA, July 6-11 2025
Teaching
General chemistry (BSc seminar)
- Compounds, solutions, colligative properties
- Gases
- Stoichiometry
- Chemical equilibrium
- Electrochemistry
Physical chemistry 1 (BSc seminar)
- Thermodynamics
- Electrochemistry
- Reaction kinetics
- Colloid chemistry
Physical chemistry 2 (BSc lecture)
- Radiochemistry
- Nuclear structure and radioactive decomposition
- Basics of quantum mechanics and the electronic structure of atoms
- History and axioms of quantum mechanics
- Analytical solution of the Schrödinger equation: Model problems
- Numerical solution of the Schrödinger equation: Variational principle and method
- Born−Oppenheimer approximation
- Description of hydrogen-like atoms
- Description of many-electron atoms
- Electronic structure of molecules
- Symmetry, point-group theory
- LCAO-MO theory
- Hückel approach
- Applied electronic structure computations
- Basics of spectroscopy
- Rotational-vibrational spectra
- Electron spectroscopy
- Magnetic spectroscopies
- NMR and ESR
Physical chemistry 2 (BSc seminar)
- Radiochemistry
- Basics of quantum mechanics and the electronic structure of atoms
- Electronic structure of molecules
- Basics of spectroscopy
- Magnetic spectroscopies
Numerical methods for the description of molecular motions (MSc lecture)
- Basics of mathematics
- Basics of physics
- Basics of programming
- Description of molecular motions
- Dynamics of chemical reactions
Numerical methods for the description of molecular motions (MSc computer class)
- Basics of Linux
- Basics of programming
- Coding numerical methods
- Molecular rotations
- Dynamics of chemical reactions
Basics of quantum and computational chemistry (BSc lecture)
- Group theory
- Basics of classical and quantum mechanics
- Approximations of quantum chemistry (The Born-Oppenheimer approximation)
- Electronic structure computations
- Ab initio methods (HF, MP2, CI, and CC)
- DFT methods
- Bases
- Reaction dynamics
- Quasi-classical trajectory method
- Chemical graph theory and the QSPR/QSAR-modeling (Gyula Tasi)
Basics of quantum and computational chemistry (BSc computer class)
- Basics of Linux and Fortran
- Use of the Gaussian electronic structure program package
- HF, MP2, CCSD(T), and DFT methods in practice
- Determination of equilibrium structures and harmonic vibrational frequencies
- Computation of reaction heats
- Reaction dynamics
- Reaction probability computations
- Chemical graph theory, linear fitting, and QSAR-modeling in practice (Gyula Tasi)
Computer modeling (in the material and medicine research) (MSc lecture)
- Formalisms and limits of classical mechanics
- Newton, Lagrange, and Hamilton formalisms
- Basics of the special relativity theory
- Postulates of quantum mechanics
- The Born-Oppenheimer approximation and beyond
- Basics of the variational and perturbational methods
- Electronic structure computation
- HF, MP2, CI, CC, and MRCI ab initio methods
- Spin-orbit couplings
- Potential energy surfaces
- Nuclear motion computations
- Rotation and vibration (anharmonic vibration and vibration-rotation coupling)
- Reaction dynamics
- Quasi-classical trajectory method
- Basics of quantum dynamics
Basics of molecular structure theory (MSc lecture)
- Introduction, historical overview
- The VSEPR theory and group theory (point groups)
- History and postulates of quantum mechanics
- Electronic structure of atoms
- H atom, quantum numbers
- Many electronic atoms
- Electronic structure of molecules
- LCAO-MO theory
- Applied electronic structure computations
- Orbital, orbital energy, electron correlation
- Electronic structure methods
Advanced chemistry - physical chemistry (MSc lecture)
- Basics of mathematics and physics, molecular structure and symmetry
- Introduction to spectroscopy
- Molecular rotations
- Molecular vibrations
- Electron spectroscopy
- Nuclear magnetic resonance (NMR)
- Reaction dynamics
Dynamics of molecular vibrations and chemical reactions (PhD lecture)
- The Born-Oppenheimer approximation
- Basics of the variational and perturbational methods
- Potential energy surfaces
- Variational-based solution of the rotational-vibrational Schrödinger equation
- Coordinates
- Operators (kinetic and potential energy)
- Basis functions
- Matrix representations (Finite basis representation (FBR), discrete variable representation (DVR), variational basis representation (VBR))
- Diagonalization of the Hamiltonian matrix
- Diatomic molecules (a ro-vibrational DVR code step by step)
- Reaction dynamics
- Quasi-classical trajectory (QCT) method
- Initial conditions
- Product analysis (standard methods and new developments (1GB))
- The zero-point energy problem in classical dynamics (active and passive constraints)
- Applications (mode-selective chemistry)
Media appearances
Dr. Dóra Papp and Dr. Gábor Czakó described the quantum tunneling effect of a chemical reaction in a Nature Communications article (in Hungarian)
On the homepage of SZTE, apropos of the appearance of our Nature Communications article (2025)
Featured Lendület Researcher: Gábor Czakó
On the homepage of MTA, apropos of receiving another Lendület grant (2025)
Our group on Szeged Television (in Hungarian)
Szeged TV Quantum (from 7:43), apropos of receiving another Lendület grant (2024)
Quantum effects on the atomic terrain table - Dr. Gábor Czakó won his second MTA Lendület grant (in Hungarian)
On the homepage of SZTE, apropos of receiving another Lendület grant (2024)
Dr. Gábor Czakó (University of Szeged) associate professor: "Computational chemistry can be more accurate than experiment" (in Hungarian)
On the homepage of SZTE, apropos of the Publication of the Year Prize (2022)
Theoretical reaction dynamics research at the University of Szeged (in Hungarian)
About our research group in the Journal of the Hungarian Chemical Society (Magyar Kémikusok Lapja) (2022) Cover
Momentum researchers first identify the experimental fingerprint of a chemical reaction (in Hungarian)
On the homepage of MTA, apropos of the appearance of our 3rd Nature Chemistry article (2021)
Researchers at Szeged publish an article about the chemical reaction of a complex system (in Hungarian)
On Delmagyar.hu, apropos of the appearance of our 3rd Nature Chemistry article (2021)
Chemistry without chemicals – New Nature Chemistry article from the SZTE Czakó group (in Hungarian)
On the homepage of SZTE, apropos of the appearance of our 3rd Nature Chemistry article (2021)
Momentum: six researchers won support in Szeged (in Hungarian)
On Delmagyar.hu, introduction of the Momentum groups of our Department (2021)
The Department of Physical Chemistry and Materials Science at the University of Szeged is the most "Momentous" (in Hungarian)
On the homepage of SZTE, introduction of the Momentum groups of our Department (2021)
"Bolyaisok" – Gábor Czakó theoretical chemist (in Hungarian)
On the homepage of MTA, showcasing the most successful researchers supported by the Bolyai Scholarship of the Hungarian Academy of Sciences (2021)
University of Szeged gives prizes to its excellent teachers and researchers (in Hungarian)
On the homepage of SZTE, apropos of receiving the Young Researcher of the Year certificate of merit (2020)
Our group on Szeged Television (in Hungarian)
Szeged TV Quantum (from 7:15), apropos of receiving the Lendület grant (2019)
Energy efficiency research in Szeged: Group of Gábor Czakó may join the international frontline (in Hungarian)
In Délmagyarország, apropos of receiving the Lendület grant (2019)
Gábor Czakó receives Momentum research grant (in English)
On the homepage of Emory University, apropos of receiving the Momentum (Lendület) grant (2019)
Two researchers of the University of Szeged, Gábor Czakó and Éva Anna Enyedy won at the MTA Lendület program in 2019 (in Hungarian)
On the homepage of SZTE, apropos of receiving the Lendület grant (2019)
From the medieval music records to the risk prediction of the coronary artery disease – the new winners of the MTA Lendület program (in Hungarian)
On the homepage of the Hungarian Academy of Sciences, apropos of receiving the Lendület grant (2019)
Collaboration of theory and experiment (in Hungarian)
Interview about our research in the Journal of the Hungarian Chemical Society (Magyar Kémikusok Lapja) (2019)
Publishing and programming (in Hungarian)
In CAMPUS Plusz (eduline), about our research group in a HVG magazine (2018)
Breaking the bond: To take part or not? (in English)
On ScienceDaily, apropos of the appearance of a Science Advances article (2018)
Theoretical chemistry and non-linear dynamics research in the Institute of Chemistry of the University of Szeged (in Hungarian)
About our research group in the Journal of the Hungarian Chemical Society (Magyar Kémikusok Lapja) (2018)
Gábor Czakó, theoretical chemist: "Unpublished results of a researcher simply do not exist" (in Hungarian)
On the homepage of SZTE, apropos of the appearance of a new Nature Chemistry article (2017)
People read about SZTE in the USA! (in Hungarian)
On SZEGEDMA online, apropos of the appearance of a Feature Article in J. Phys. Chem. A (2017)
Study clarifies the role of the leaving group in gas-phase bimolecular reaction (in English)
On phys.org, apropos of the appearance of the group's first Nature Chemistry article (2015)
Chemists from the University of Szeged in the world-leading chemistry journal (in Hungarian)
On SZEGEDMA online, apropos of the appearance of the group's first Nature Chemistry article (2015)
Groundbreaking discovery of chemists from the University of Szeged (in Hungarian)
In Délmagyarország, apropos of the appearance of the group's first Nature Chemistry article (2015)
Achievements of chemists from the University of Szeged highlighted in the world-leading chemistry journal (in Hungarian)
On the homepage of SZTE, apropos of the appearance of the group's first Nature Chemistry article (2015)
Double inversion live on Hungarian Television (in Hungarian)
MTVA Ma reggel (from 2:36:13), apropos of the discovery of a new reaction pathway published in Nature Communications (2015)
Groundbreaking discovery of Hungarian chemists (in Hungarian)
Index.hu, apropos of the discovery of a new reaction pathway published in Nature Communications (2015)
Discovery of a new reaction pathway (in Hungarian)
National Geographic Hungary, apropos of the discovery of a new double-inversion mechanism for SN2 reactions published in Nature Communications (2015)
The chemist Gábor Czakó received the Junior Prima Prize (in Hungarian)
Délmagyar (2012)
Molecular modeler (in Hungarian)
Figyelő, interview after returning home from the United States (2012)
Theoretical chemists find new dimension to rules for reactions (in English)
phys.org, apropos of the Science paper and the modification of the Polanyi rules (2011)
Gábor Czakó featured by the word-famous magazine (in Hungarian)
Délmagyar, apropos of the Science highlight of the F + methane paper [J. Am. Chem. Soc. 131, 17534 (2009)] (2010)