About the CRD research group
|
Current members: 20 Publications: 98 Nature Chemistry papers: 3 PhD dissertations: 9 MSc theses: 9 BSc theses: 24 MSc project work: 6 Undergraduate research reports: 16 |

|
Members
Head of group
Dr. Gábor CzakóProfessor, Doctor of the Hungarian Academy of Sciences
Department of Physical Chemistry and Materials Science, University of Szeged
Rerrich Béla tér 1, H-6720 Szeged, Hungary
Office: BE-417, Phone: +36-62-34-3742
E-mail: gczako at chem.u-szeged.hu
External members
 Dr. Dóra Papp
Dr. Dóra PappAssistant professor
Postdoc in the group (2018-2024)
Office: BE-420
E-mail: dorapapp at chem.u-szeged.hu
Homepage
Postdocs

Dr. Attila Dékány
Postdoc (2025 - ), Predoc (2024-2025), PhD student (2020-2024)
Office: BE-418
E-mail: dekanyattilaadam at gmail.com

Dr. Balázs Gruber
Postdoc (2025 - ), Predoc (2024-2025), PhD student (2020-2024)
MSc student (2018-2020), BSc student (2017-2018)
E-mail: gbalazs96 at gmail.com

Dr. Tibor Győri
Postdoc (2025 - ), Predoc (2022-2025), PhD student (2018-2022)
MSc student (2016-2018), BSc student (2015-2016)
Office: BE-414
E-mail: tiborgyri at gmail.com

Dr. Viktor Tajti
Postdoc (2024 - ), Predoc (2023-2024), PhD student (2019-2023)
MSc student (2017-2019), BSc student (2016-2017)
Office: BE-414
E-mail: vtajti at chem.u-szeged.hu
PhD students

Petra Tóth
PhD student (2024 - ), MSc student (2022-2024), BSc student (2020-2022)
Office: BE-414
E-mail: toth.petra.294 at gmail.com
Research associates

Dr. András Nacsa
Research associate (2024 - ), Predoc (2024), PhD student (2020-2024)
Research associate (2018-2020), BSc student (2016-2018)
E-mail: nacsa.andras.bence at gmail.com

Dr. Tímea Szűcs
Research associate (2025 - ), Predoc (2024-2025), PhD student (2020-2024), Research associate (2020)
E-mail: szucsmeja at gmail.com
MSc students

Bálint Imre
MSc student (2024 - ), BSc student (2023-2024)
Office: BE-418
E-mail: balintimre0902 at gmail.com

Balázs Molnár
MSc student (2025 - ), BSc student (2022-2025)
E-mail: 2017molbal at gmail.com

Csaba Rudner
MSc student (2024 - ), BSc student (2023-2024)
Office: BE-418
E-mail: rudnercsaba02 at gmail.com

Levente Sárik
(2025 - )
E-mail: levi200320 at gmail.com
BSc students

Anna Bohr
(2025 - )
E-mail: bohrpanka at gmail.com

Márton Felföldi
(2024 - )
E-mail: bestfmc5 at gmail.com

István Jancsó
(2025 - )
E-mail: jisti88 at gmail.com

Lili Kószó
(2025 - )
E-mail: koszolili10 at gmail.com

Panka Viola Péter
(2025 - )
E-mail: peterpanka.school at gmail.com

Noémi Szabadszállási
(2025 - )
E-mail: szabadszallasinoemi at gmail.com

Dominika Takó
BSc student (2025 - ), High-school research student (2023-2025)
E-mail: tako.dominika at gmail.com
Former members

Boldizsár Ballay
BSc student (2022-2023)

Bartal Baranyi
High-school research student (2023)

Laura Erdei
MSc student, project work (2024-2025), BSc student (2023-2024)

Zita Fábián
MSc student, project work (2017)

Péter Fuder
High-school research student (2023)

Dorina Gál
Research associate (2024-2025), BSc student (2023-2024)

Anett Giricz
BSc student (2020-2022)

Réka Gyimesi
Research associate (2024-2025), BSc student (2023-2024)

Bálint Hajdu
MSc student, project work (2016)

Kitti Horváth
MSc student (2023-2025), BSc student (2021-2023)

Zsolt Kerekes
MSc student (2021-2023), BSc student (2019-2021)

Máté Kígyósi
BSc student (2021-2022)

Nándor Kocsis
High-school research student (2023)

Gyula Kovács
BSc student (2016-2017)

László Krotos
MSc student, project work (2017)

Anett Major
BSc student (2024-2025)

Dr. Balázs Olasz
PhD student (2015-2019)

Erik Orján
MSc student (2018-2020), BSc student (2017-2018)

Paszkál Papp
MSc student, project work (2020)

Anna Katinka Schmidt
BSc student (2019-2020)

Barnabás Sipos
Research associate (2022-2023), BSc student (2020-2022)

Dr. István Szabó
PhD student (2012-2016)

Gergő Péter Szekeres
MSc student, project work (2016)

Dorina Szepesi
High-school research student (2020)

Dr. Domonkos Attila Tasi
Postdoc (2023-2024), Predoc (2021-2023), PhD student (2017-2021)
Homepage

Csenge Tokaji
MSc student (2021-2024), BSc student (2019-2021)

Adrián Traj
MSc student, project work (2020)

Demeter Ván
MSc student (2019-2021), BSc student (2017-2019)

Dr. Cangtao Yin
Postdoc (2022-2024)
Research
Benchmark ab initio thermochemistry
We employ modern electronic structure methods to determine the best technically feasible stationary-point properties, such as structures, relative energies, harmonic vibrational frequencies, for reactive chemical systems. The computed results provide benchmark thermochemical data and guide future experimental and theoretical investigations. Furthermore, the characterization of the stationary points is the first step toward developing potential energy surfaces for reaction dynamics studies.
Potential energy surface developments
Potential energy surfaces (PESs) govern the motions of the atoms in a chemical reaction. We develop analytical global PESs for reactive chemical systems by fitting high-level ab initio energy points. Our PESs allow efficient dynamical investigations usually with unprecedented detail and accuracy. We have accurate full-dimensional PESs for the X + CH4 [X = F, O, Cl, Br] and F− + CH3Y [Y = F, Cl, I] reactions and we are currently working on PES developments for several interesting chemical systems up to 12 atoms.
Reaction dynamics computations
With analytical PESs at hand we can study the reaction dynamics using the quasi-classical trajectory and/or, in collaboration with leading Chinese and American groups, quantum mechanical methods. The reaction dynamics computations follow the motions of the atoms step by step, thereby revealing novel reaction pathways and different outcomes of a chemical reaction. Furthermore, our simulations can help and motivate experimental investigations and resulted in active collaborations with top experimental research groups in Austria and Taiwan.
Dynamics of the reactions of atoms and radicals with alkanes
We have studied the dynamics of the reactions of methane with the F, O, Cl, and Br atoms leading to many groundbreaking discoveries such as the modifications of the rules of Nobel-laureate John Polanyi for polyatomic systems, vibrational and rotational mode specificity, angle-dependent barrier, novel reaction pathways, etc. Some of these findings were reported in the best scientific journals of the world such as Science, Nature Chemistry, PNAS, and JACS.
Dynamics of SN2 reactions
Bimolecular nucleophilic substitution (SN2) reactions play a central role in chemistry and biochemistry and their Walden-inversion mechanism has been known for more than a hundred years. Recently we initiated a novel way to study the dynamics of these SN2 reactions based on analytical PESs. Our simulations revealed (a) a new mechanisms, called double inversion, for SN2 reactions, (b) unexpected leaving group effects, (c) non-traditional dynamics involving front-side complex formation, (d) unprecedented agreement with experiment, etc. These results were published in high-impact journals, Nature Communications, Nature Chemistry, and Chemical Science, and were featured in the national and international media and on the front cover of the Journal of Physical Chemistry A.
Dynamics of small biosystems
We are currently working on novel techniques to represent PESs of small biosystems such as amino acids, thereby allowing the study of their conformational dynamics and reactions with atoms and radicals. Our simulations are expected to be more accurate and reliable than previous work that used classical force fields to represent PESs.
Publications since August of 2015
I. Szabó and G. Czakó
Rotational mode specificity in the F− + CH3Y [Y = F and Cl] SN2 reactions
J. Phys. Chem. A 119, 12231 (2015) PDF
M. Stei, E. Carrascosa, M. A. Kainz, A. H. Kelkar, J. Meyer, I. Szabó, G. Czakó, and R. Wester
Influence of the leaving group on the dynamics of a gas-phase SN2 reaction
Nat. Chem. 8, 151 (2016) PDF Highly Cited Paper (as of July/August 2016)
Y. Wang, H. Song, I. Szabó, G. Czakó, H. Guo, and M. Yang
Mode-specific SN2 reaction dynamics
J. Phys. Chem. Lett. 7, 3322 (2016) PDF
I. Szabó and G. Czakó
Mode-specific multi-channel dynamics of the F− + CHD2Cl reaction on a global ab initio potential energy surface
J. Chem. Phys. 145, 134303 (2016) PDF
B. Olasz, I. Szabó, and G. Czakó
High-level ab initio potential energy surface and dynamics of the F− + CH3I SN2 and proton-transfer reactions
Chem. Sci. 8, 3164 (2017) PDF
V. Tajti and G. Czakó
Benchmark ab initio characterization of the complex potential energy surface of the F− + CH3CH2Cl reaction
J. Phys. Chem. A 121, 2847 (2017) PDF
I. Szabó, B. Olasz, and G. Czakó
Deciphering front-side complex formation in SN2 reactions via dynamics mapping
J. Phys. Chem. Lett. 8, 2917 (2017) PDF
I. Szabó and G. Czakó
Benchmark ab initio characterization of the complex potential energy surface of the Cl− + CH3I reaction
J. Phys. Chem. A 121, 5748 (2017) PDF
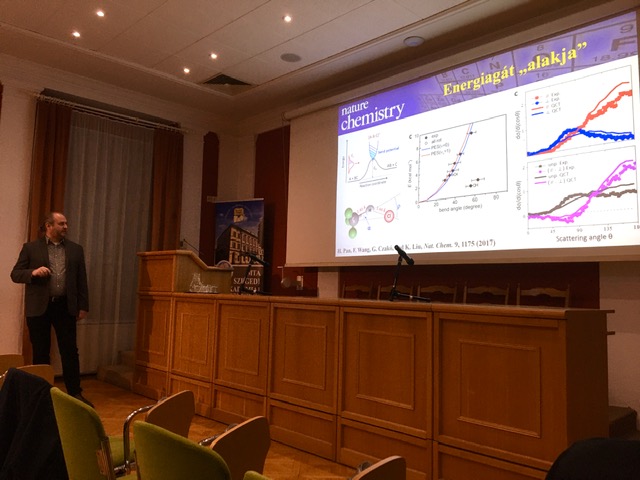
H. Pan, F. Wang, G. Czakó, and K. Liu
Direct mapping of the angle-dependent barrier to reaction for Cl + CHD3 using polarized scattering data
Nat. Chem. 9, 1175 (2017) PDF
I. Szabó and G. Czakó
Dynamics and novel mechanisms of SN2 reactions on ab initio analytical potential energy surfaces
J. Phys. Chem. A 121, 9005 (2017) Feature Article Cover PDF
L. Krotos and G. Czakó
Does the Cl + CH4 __> H + CH3Cl reaction proceed via Walden inversion?
J. Phys. Chem. A 121, 9415 (2017) PDF
B. Hajdu and G. Czakó
Benchmark ab initio characterization of the complex potential energy surfaces of the X− + NH2Y [X,Y = F, Cl, Br, I] reactions
J. Phys. Chem. A 122, 1886 (2018) PDF
T. Győri, B. Olasz, G. Paragi, and G. Czakó
Effects of the level of electronic structure theory on the dynamics of the F− + CH3I reaction
J. Phys. Chem. A 122, 3353 (2018) PDF
S. Góger, P. Szabó, G. Czakó, and G. Lendvay
Flame inhibition chemistry: rate coefficients of the reactions of HBr with CH3 and OH radicals at high temperatures determined by quasiclassical trajectory calculations
Energy Fuels 32, 10100 (2018) PDF
M. Stei, E. Carrascosa, A. Dörfler, J. Meyer, B. Olasz, G. Czakó, A. Li, H. Guo, and R. Wester
Stretching vibration is spectator in nucleophilic substitution
Sci. Adv. 4, eaas9544 (2018) PDF
D. A. Tasi, Z. Fábián, and G. Czakó
Benchmark ab initio characterization of the inversion and retention pathways of the OH− + CH3Y [Y = F, Cl, Br, I] SN2 reactions
J. Phys. Chem. A 122, 5773 (2018) PDF
B. Olasz and G. Czakó
Mode-specific quasiclassical dynamics of the F− + CH3I SN2 and proton-transfer reactions
J. Phys. Chem. A 122, 8143 (2018) PDF
D. Papp, B. Gruber, and G. Czakó
Detailed benchmark ab initio mapping of the potential energy surfaces of the X + C2H6 [X = F, Cl, Br, I] reactions
Phys. Chem. Chem. Phys. 21, 396 (2019) PDF
B. Bastian, E. Carrascosa, A. Kaiser, J. Meyer, T. Michaelsen, G. Czakó, W. L. Hase, and R. Wester
Dynamics of proton transfer from ArH+ to CO
Int. J. Mass Spectrom. 438, 175 (2019) PDF
B. Olasz and G. Czakó
High-level-optimized stationary points for the F−(H2O) + CH3I system: Proposing a new water-induced double-inversion pathway
J. Phys. Chem. A 123, 454 (2019) PDF
B. Olasz and G. Czakó
Uncovering the role of the stationary points in the dynamics of the F− + CH3I reaction
Phys. Chem. Chem. Phys. 21, 1578 (2019) PDF
D. A. Tasi, Z. Fábián, and G. Czakó
Rethinking the X− + CH3Y [X = OH, SH, CN, NH2, PH2; Y = F, Cl, Br, I] SN2 reactions
Phys. Chem. Chem. Phys. 21, 7924 (2019) PDF
G. Czakó
Dynamics and mechanisms of fundamental chemical reactions
Magy. Kém. Foly. 125, 100 (2019) (in Hungarian) PDF
G. Czakó, T. Győri, B. Olasz, D. Papp, I. Szabó, V. Tajti, and D. A. Tasi
Benchmark ab initio and dynamical characterization of the stationary points of reactive atom + alkane and SN2 potential energy surfaces
Phys. Chem. Chem. Phys. 22, 4298 (2020) Perspective Cover PDF
T. Győri and G. Czakó
Automating the development of high-dimensional reactive potential energy surfaces with the ROBOSURFER program system
J. Chem. Theory Comput. 16, 51 (2020) PDF Highly Cited Paper (as of May/June 2020)
G. Avila, D. Papp, G. Czakó, and E. Mátyus
Exact quantum dynamics background of dispersion interactions: case study for CH4.Ar in full (12) dimensions
Phys. Chem. Chem. Phys. 22, 2792 (2020) PDF Selected as a 2020 HOT PCCP article
D. A. Tasi, T. Győri, and G. Czakó
On the development of a gold-standard potential energy surface for the OH− + CH3I reaction
Phys. Chem. Chem. Phys. 22, 3775 (2020) Communication PDF Selected as a 2020 HOT PCCP article
D. Papp, V. Tajti, T. Győri, and G. Czakó
Theory finally agrees with experiment for the dynamics of the Cl + C2H6 reaction
J. Phys. Chem. Lett. 11, 4762 (2020) PDF Publication of the Month (Chemical Sciences Section, Hungarian Academy of Sciences)
E. M. Orján, A. B. Nacsa, and G. Czakó
Conformers of dehydrogenated glycine isomers
J. Comput. Chem. 41, 2001 (2020) PDF
B. Gruber and G. Czakó
Benchmark ab initio characterization of the abstraction and substitution pathways of the OH + CH4/C2H6 reactions
Phys. Chem. Chem. Phys. 22, 14560 (2020) PDF
P. Papp, V. Tajti, and G. Czakó
Numerical separation of the front-side attack and double-inversion retention pathways of SN2 reactions
Chem. Phys. Lett. 755, 137780 (2020) PDF
D. Papp and G. Czakó
Full-dimensional MRCI-F12 potential energy surface and dynamics of the F(2P3/2) + C2H6 __> HF + C2H5 reaction
J. Chem. Phys. 153, 064305 (2020) PDF
P. Papp and G. Czakó
Rotational mode specificity in the F− + CH3I(v=0, JK) SN2 and proton-transfer reactions
J. Phys. Chem. A 124, 8943 (2020) PDF
G. Czakó, T. Győri, D. Papp, V. Tajti, and D. A. Tasi
First-principles reaction dynamics beyond six-atom systems
J. Phys. Chem. A 125, 2385 (2021) Perspective Cover PDF
D. Papp and G. Czakó
Facilitated inversion complicates the stereodynamics of an SN2 reaction at nitrogen center
Chem. Sci. 12, 5410 (2021) Cover PDF
A. B. Nacsa and G. Czakó
Benchmark ab initio proton affinity of glycine
Phys. Chem. Chem. Phys. 23, 9663 (2021) Cover PDF
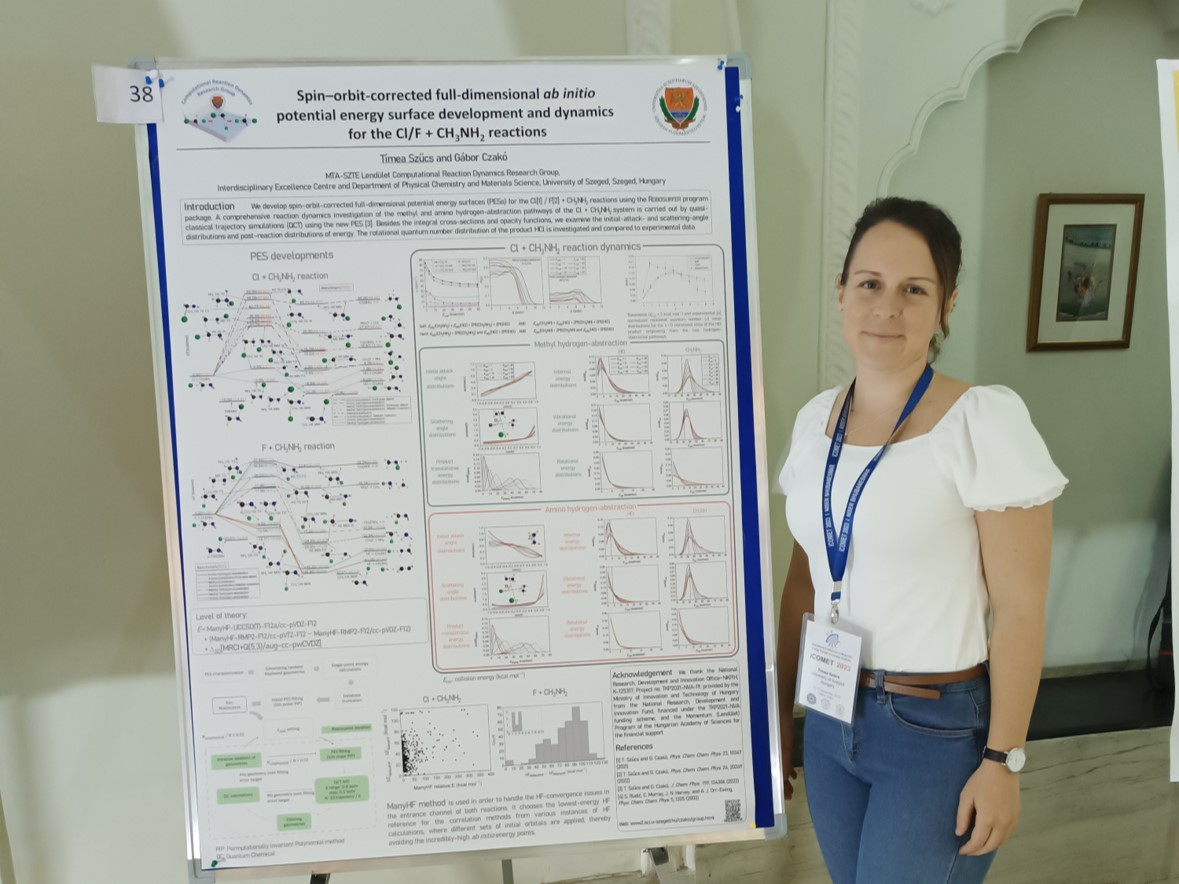
T. Szűcs and G. Czakó
Benchmark ab initio stationary-point characterization of the complex potential energy surface of the multi-channel Cl + CH3NH2 reaction
Phys. Chem. Chem. Phys. 23, 10347 (2021) PDF
D. A. Tasi, C. Tokaji, and G. Czakó
A benchmark ab initio study of the complex potential energy surfaces of the OH− + CH3CH2Y [Y = F, Cl, Br, I] reactions
Phys. Chem. Chem. Phys. 23, 13526 (2021) PDF Selected as a 2021 HOT PCCP article
J. Meyer, V. Tajti, E. Carrascosa, T. Győri, M. Stei, T. Michaelsen, B. Bastian, G. Czakó, and R. Wester
Atomistic dynamics of elimination and nucleophilic substitution disentangled for the F− + CH3CH2Cl reaction
Nat. Chem. 13, 977 (2021) PDF Publication of the Month (Chemical Sciences Section, Hungarian Academy of Sciences)
Publication of the Year (University of Szeged)
D. Papp, J. Li, H. Guo, and G. Czakó
Vibrational mode-specificity in the dynamics of the Cl + C2H6 __> HCl + C2H5 reaction
J. Chem. Phys. 155, 114303 (2021) PDF
V. Tajti, T. Győri, and G. Czakó
Detailed quasiclassical dynamics of the F− + CH3Br reaction on an ab initio analytical potential energy surface
J. Chem. Phys. 155, 124301 (2021) PDF
D. Papp and G. Czakó
Vibrational mode-specific dynamics of the F(2P3/2) + C2H6 __> HF + C2H5 reaction
J. Chem. Phys. 155, 154302 (2021) PDF
D. A. Tasi and G. Czakó
Uncovering an oxide ion substitution for the OH− + CH3F reaction
Chem. Sci. 12, 14369 (2021) Cover PDF Selected for the 2021 Chemical Science HOT Article Collection
A. Á. Dékány, G. Z. Kovács, and G. Czakó
High-level systematic ab initio comparison of carbon- and silicon-centered SN2 reactions
J. Phys. Chem. A 125, 9645 (2021) PDF
A. Á. Dékány and G. Czakó
Benchmark ab initio proton affinity and gas-phase basicity of α-alanine based on coupled-cluster theory and statistical mechanics
J. Comput. Chem. 43, 19 (2022) PDF
Z. Kerekes, D. A. Tasi, and G. Czakó
SN2 reactions with an ambident nucleophile: A benchmark ab initio study of the CN− + CH3Y [Y = F, Cl, Br, and I] systems
J. Phys. Chem. A 126, 889 (2022) PDF
T. Győri and G. Czakó
ManyHF: A pragmatic automated method of finding lower-energy Hartree−Fock solutions for potential energy surface development
J. Chem. Phys. 156, 071101 (2022) Communication Cover PDF Selected as Featured
V. Tajti and G. Czakó
Vibrational mode-specific dynamics of the F− + CH3CH2Cl multi-channel reaction
Phys. Chem. Chem. Phys. 24, 8166 (2022) PDF
D. Papp and G. Czakó
Rotational mode-specificity in the Cl + C2H6 __> HCl + C2H5 reaction
J. Phys. Chem. A 126, 2551 (2022) PDF
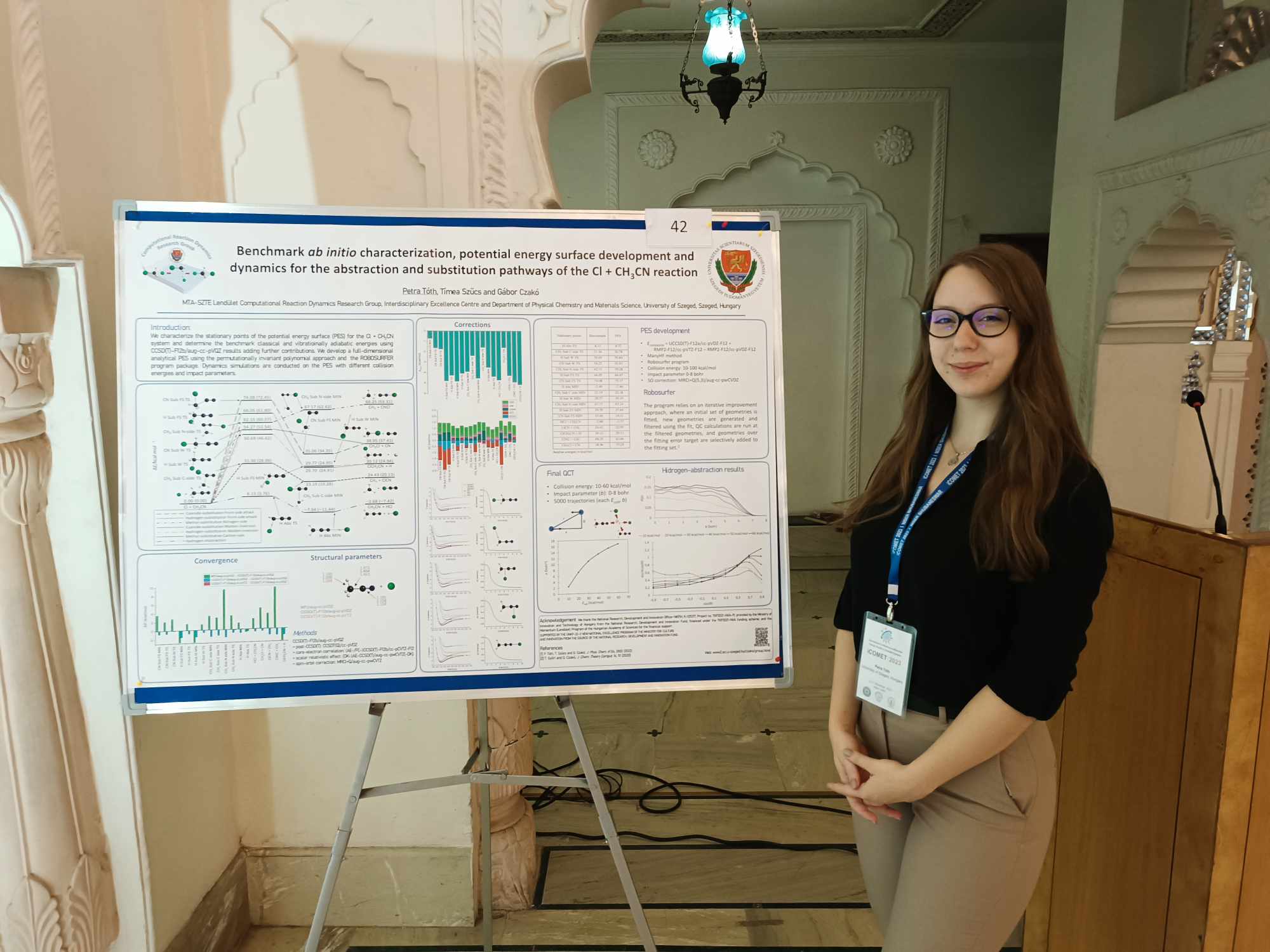
P. Tóth, T. Szűcs, and G. Czakó
Benchmark ab initio characterization of the abstraction and substitution pathways of the Cl + CH3CN reaction
J. Phys. Chem. A 126, 2802 (2022) PDF
D. A. Tasi and G. Czakó
Unconventional SN2 retention pathways induced by complex formation: High-level dynamics investigation of the NH2− + CH3I polyatomic reaction
J. Chem. Phys. 156, 184306 (2022) PDF
B. Gruber, V. Tajti, and G. Czakó
Full-dimensional automated potential energy surface development and dynamics for the OH + C2H6 reaction
J. Chem. Phys. 157, 074307 (2022) PDF
T. Szűcs and G. Czakó
Benchmark ab initio potential energy surface mapping of the F + CH3NH2 reaction
Phys. Chem. Chem. Phys. 24, 20249 (2022) PDF
C. Yin, V. Tajti, and G. Czakó
Full-dimensional potential energy surface development and dynamics for the HBr + C2H5 __> Br(2P3/2) + C2H6 reaction
Phys. Chem. Chem. Phys. 24, 24784 (2022) PDF Selected as a 2022 HOT PCCP article
C. Yin and G. Czakó
Automated full-dimensional potential energy surface development and quasi-classical dynamics for the HI(X1Σ+) + C2H5 __> I(2P3/2) + C2H6 reaction
Phys. Chem. Chem. Phys. 24, 29084 (2022) PDF
A. B. Nacsa and G. Czakó
Benchmark ab initio determination of the conformers, proton affinities, and gas-phase basicities of cysteine
J. Phys. Chem. A 126, 9667 (2022) PDF
D. Papp, V. Tajti, G. Avila, E. Mátyus, and G. Czakó
CH4.F− revisited: full-dimensional ab initio potential energy surface and variational vibrational states
Mol. Phys. 121, e2113565 (2023) PDF
C. Yin and G. Czakó
Theoretical vibrational mode-specific dynamics studies for the HBr + C2H5 reaction
Phys. Chem. Chem. Phys. 25, 3083 (2023) PDF
D. A. Tasi, T. Michaelsen, R. Wester, and G. Czakó
Quasi-classical trajectory study of the OH− + CH3I reaction: Theory meets experiment
Phys. Chem. Chem. Phys. 25, 4005 (2023) PDF
T. Győri and G. Czakó
A comprehensive benchmark ab initio survey of the stationary points and products of the OH· + CH3OH system
J. Chem. Phys. 158, 034301 (2023) PDF
B. Gruber and G. Czakó
High-level ab initio mapping of the multiple H-abstraction pathways of the OH + glycine reaction
Phys. Chem. Chem. Phys. 25, 5271 (2023) PDF
A. B. Nacsa, M. Kígyósi, and G. Czakó
Protonation of serine: Conformers, proton affinities and gas-phase basicities at the "gold standard" and beyond
Phys. Chem. Chem. Phys. 25, 8891 (2023) PDF
C. Yin and G. Czakó
Vibrational mode-specific quasi-classical trajectory studies for the two-channel HI + C2H5 reaction
Phys. Chem. Chem. Phys. 25, 9944 (2023) PDF
A. B. Nacsa, V. Tajti, and G. Czakó
Dynamics of the Cl− + CH3I reaction on a high-level ab initio analytical potential energy surface
J. Chem. Phys. 158, 194306 (2023) PDF
A. Á. Dékány and G. Czakó
Exploring the versatile reactivity of the F− + SiH3Cl system on a full-dimensional coupled-cluster potential energy surface
J. Chem. Phys. 158, 224303 (2023) PDF
T. Gstir, T. Michaelsen, B. A. Long, A. B. Nacsa, A. Ayasli, D. Swaraj, F. Zappa, F. Trummer, S. G. Ard, N. S. Shuman, G. Czakó, A. A. Viggiano, and R. Wester
The influence of fluorination on the dynamics of the F− + CH3CH2I reaction
Phys. Chem. Chem. Phys. 25, 18711 (2023) PDF Selected as a 2023 HOT PCCP article
C. Yin and G. Czakó
Competition between the H-abstraction and the X-abstraction pathways in the HX (X = Br, I) + C2H5 reactions
Phys. Chem. Chem. Phys. 25, 20241 (2023) PDF Selected as a 2023 HOT PCCP article
A. Giricz, G. Czakó, and D. Papp
Alternating stereospecificity upon central-atom change: Dynamics of the F− + PH2Cl SN2 reaction compared to its C- and N-centered analogues
Chem. Eur. J. 29, e202302113 (2023) PDF Publication of the Month (Chemical Sciences Section, Hungarian Academy of Sciences)
B. Gruber, V. Tajti, and G. Czakó
Vibrational mode-specific dynamics of the OH + C2H6 reaction
J. Phys. Chem. A 127, 7364 (2023) PDF
T. Szűcs and G. Czakó
ManyHF-based full-dimensional potential energy surface development and quasi-classical dynamics for the Cl + CH3NH2 reaction
J. Chem. Phys. 159, 134306 (2023) PDF
C. Yin and G. Czakó
Full-dimensional automated potential energy surface development and detailed dynamics for the CH2OO + NH3 reaction
Phys. Chem. Chem. Phys. 25, 26917 (2023) PDF
B. Ballay, T. Szűcs, D. Papp, and G. Czakó
Phosphorus-centered ion-molecule reactions: benchmark ab initio characterization of the potential energy surfaces of the X− + PH2Y [X, Y = F, Cl, Br, I] systems
Phys. Chem. Chem. Phys. 25, 28925 (2023) PDF
D. A. Tasi and G. Czakó
Vibrational mode-specificity in the dynamics of the OH− + CH3I multi-channel reaction
J. Chem. Phys. 160, 044305 (2024) PDF
T. Szűcs and G. Czakó
Automated potential energy surface development and comprehensive dynamics for the F + CH3NH2 reaction
J. Chem. Phys. 160, 064304 (2024) PDF Editor's Pick
A. B. Nacsa, C. Tokaji, and G. Czakó
High-level analytical potential-energy-surface-based dynamics of the OH− + CH3CH2Cl SN2 and E2 reactions in full (24) dimensions
Faraday Discuss. 251, 604 (2024) PDF
A. Á. Dékány and G. Czakó
Detailed quasiclassical dynamics of the F− + SiH3Cl multi-channel reaction
Phys. Chem. Chem. Phys. 26, 10008 (2024) PDF
A. Ayasli, P. Tóth, T. Michaelsen, T. Gstir, F. Zappa, D. Papp, G. Czakó, and R. Wester
Imaging the ion-molecule reaction dynamics of O− + CD4
J. Phys. Chem. A 128, 3078 (2024) PDF
G. Czakó, B. Gruber, D. Papp, V. Tajti, D. A. Tasi, and C. Yin
First-principles mode-specific reaction dynamics
Phys. Chem. Chem. Phys. 26, 15818 (2024) Perspective (PCCP 25th Anniversary Collection) Cover PDF Selected as a 2024 HOT PCCP article
D. A. Tasi and G. Czakó
Benchmark ab initio characterization of the complex potential energy surfaces of the HOO− + CH3Y [Y = F, Cl, Br, I] reactions
Phys. Chem. Chem. Phys. 26, 16048 (2024) PDF
K. Horváth, V. Tajti, D. Papp, and G. Czakó
Dynamics of the HCl + C2H5 multi-channel reaction on a full-dimensional ab initio potential energy surface
J. Phys. Chem. A 128, 4474 (2024) PDF
D. R. Gál, D. Papp, and G. Czakó
Benchmark ab initio characterization of the multi-channel Cl + CH3X [X = F, Cl, Br, I] reactive potential energy surfaces
Phys. Chem. Chem. Phys. 26, 17695 (2024) PDF
C. Yin and G. Czakó
Revealing new pathways for the reaction of Criegee intermediate CH2OO with SO2
Commun. Chem. 7, 157 (2024) PDF
P. Tóth, T. Szűcs, T. Győri, and G. Czakó
Dynamics of the Cl + CH3CN reaction on an automatically-developed full-dimensional ab initio potential energy surface
J. Chem. Phys. 161, 084304 (2024) PDF
E. Mátyus and G. Czakó
Foreword to the Festschrift in honour of Professor Attila G. Császár: molecules (always) in motion
Mol. Phys. 122, e2360850 (2024) PDF
G. Alexandrowicz, D. Babikov, M. Brouard, A. Butler, H. Chadwick, D. W. Chandler, M. Fárník, J. Fingerhut, H. Guo, T. Győri, C. T. Haakansson, D. J. Harding, D. Heard, B. R. Heazlewood, D. Heathcote, N. Hertl, P. G. Jambrina, G.-J. Kroes, O. A. Krohn, P. D. Lane, V. Le Duc, H. J. Lewandowski, J. Loreau, M. McCrea, K. G. McKendrick, J. Meyer, D. R. Moon, A. S. Mullin, G. M. Nathanson, D. M. Neumark, K.-K. Ni, N. Pal, E. Pluhařová, C. Reilly, P. Robertson, S. J. Sibener, C. Sparling, V. Sridurai, A. Srivastav, M. Strutton, A. G. Suits, J. Wagner, P. D. Watson, R. Wester, S. Willitsch, A. M. Wodtke, and B. S. Zhao
Scattering in extreme environments: general discussion
Faraday Discuss. 251, 171 (2024) PDF
D. Babikov, N. Balucani, A. Bergeat, M. Brouard, D. W. Chandler, M. L. Costen, M. Fárník, H. Guo, T. Győri, D. Heard, D. Heathcote, N. Hertl, P. G. Jambrina, N. M. Kidwell, O. A. Krohn, V. Le Duc, J. Loreau, S. R. Mackenzie, M. McCrea, K. G. McKendrick, J. Meyer, D. R. Moon, A. S. Mullin, G. S. Nathanson, D. M. Neumark, K.-K. Ni, M. J. Paterson, E. Pluhařová, P. Robertson, C. Reilly, G. C. Schatz, C. Sparling, A. G. Suits, P. D. Watson, R. Wester, S. Willitsch, and A. M. Wodtke
Scattering of larger molecules – part 1: general discussion
Faraday Discuss. 251, 313 (2024) PDF
F. J. Aoiz, N. Balucani, A. Bergeat, A. Butler, D. W. Chandler, G. Czakó, T. Győri, D. E. Heard, D. Heathcote, B. R. Heazlewood, N. Hertl, P. G. Jambrina, R. I. Kaiser, O. A. Krohn, V. Le Duc, J. Loreau, S. R. Mackenzie, K. G. McKendrick, J. Meyer, G. M. Nathanson, D. M. Neumark, R. Pandey, C. Reilly, P. Robertson, G. C. Schatz, S. J. Sibener, A. G. Suits, P. D. Watson, R. Wester, S. Willitsch, A. M. Wodtke, and B. S. Zhao
Scattering of larger molecules – part 2: general discussion
Faraday Discuss. 251, 622 (2024) PDF
B. Gruber and G. Czakó
High-level ab initio characterization of the OH + CH3NH2 reaction
Phys. Chem. Chem. Phys. 26, 28543 (2024) PDF
B. J. Molnár, A. Á. Dékány, and G. Czakó
Automated potential energy surface development and quasi-classical dynamics for the F− + SiH3I system
J. Chem. Phys. 161, 194306 (2024) PDF
D. A. Tasi, E. M. Orján, and G. Czakó
Benchmark ab initio mapping of the F− + CH2ClI SN2 and proton-abstraction reactions
J. Phys. Chem. A 128, 10568 (2024) PDF
A. Ayasli, A. Khan, T. Gstir, T. Michaelsen, D. Papp, Y. Wang, H. Song, M. Yang, G. Czakó, and R. Wester
A dynamic isotope effect in the nucleophilic substitution reaction between F− and CD3I
Nat. Commun. 16, 2318 (2025) PDF
Y. Wang, Z. Tu, G. Czakó, H. Song, and M. Yang
Mode-specific quantum and quasi-classical trajectory dynamics of the F− + CH3I __> I− + CH3F SN2 reaction
J. Phys. Chem. A 129, 6344 (2025) PDF
A. Sunaga, T. Győri, G. Czakó, and E. Mátyus
Exact quantum dynamics of methanol: Full-dimensional ab initio potential energy surface of spectroscopic quality and variational vibrational states
J. Chem. Phys. 163, 064101 (2025) PDF
P. Tóth and G. Czakó
Vibrational mode-specific dynamics of the Cl + CH3CN reaction
J. Chem. Phys. 163, 064306 (2025) PDF
G. Czakó
First-principles dynamics and mechanisms of fundamental chemical reactions
Adv. Quantum Chem. 94, 43 (2026) Book chapter
B. Gruber and G. Czakó
Site- and conformer-specific reaction dynamics of glycine with the hydroxyl radical
Commun. Chem. 9, 16 (2026) PDF
A. Á. Dékány, B. J. Molnár, and G. Czakó
Mode-specific quasi-classical dynamics of the F− + SiH3Cl system
Phys. Chem. Chem. Phys. 28, 5948 (2026) PDF
L. C. Erdei, D. Papp, and G. Czakó
Leaving-group effects in SN2@P: Potential energy surface and dynamics of the F− + PH2I reaction compared to its Cl−-leaving-group analogue
ACS Phys. Chem. Au accepted (2026)
News
March 26, 2026
Laura and Dóri's paper on the PES and dynamics of the F− + PH2I reaction has been accepted by ACS Phys. Chem. Au.February 7, 2026
Attila and Balázs's mode-specific F− + SiH3Cl paper has been accepted by PCCP.January 12, 2026
FM successfully defends his BSc thesis.December 18, 2025
Anna Bohr (BSc student) joins our group.December 15, 2025
Gábor becomes Full Professor.December 6, 2025
Gábor, Balázs, and Petra give talks in Mátrafüred at the meeting of the MTA Material and Molecular Structure Working Group.November 21, 2025
Csaba gives a talk in Mátrafüred at the meeting of the MTA Reaction Kinetics and Photochemistry Working Group.November 20, 2025
Petra and Tibi give talks in Mátrafüred at the meeting of the MTA Reaction Kinetics and Photochemistry Working Group.November 20, 2025
Balázs's paper on the site- and conformer-specific dynamics of the OH + glycine reaction has been accepted by Communications Chemistry.October 3, 2025
Attila successfully defends his PhD dissertation (see Press).September 8, 2025
Levente Sárik (MSc student) and István Jancsó (BSc student) join the group.September 3, 2025
Tibi gives a contributed talk at the International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) in Szeged.September 1, 2025
Balázs Gruber, Balázs Molnár, Petra, and Viktor present their posters at the International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) (Szeged).September 1, 2025
Petra gives a hot topic talk at the International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) in Szeged.September 1, 2025
Petra wins the scholarship of the University Research Fellowship Program (EKÖP).August 31, 2025
Gábor opens the International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) held in Szeged.August 22, 2025
Gábor gives lectures at the PHYMOL Training School at Eötvös University.July 18, 2025
Petra's mode-specific Cl + CH3CN paper has been accepted by J. Chem. Phys.July 16, 2025
Tibi's paper, written in collaboration with Ayaki Sunaga and Edit Mátyus, on the PES and vibrational states of methanol has been accepted by J. Chem. Phys.July 7-8, 2025
Petra presents her poster at the Dynamics of Molecular Collisions (DMC) Conference (Snowbird, Utah, USA).June 27, 2025
Balázs, Tibi, and Timi receive their PhD certificates.June 26, 2025
Gábor's paper, written in collaboration with Yan Wang and co-workers, on the quantum and QCT dynamics of the F− + CH3I SN2 reaction has been accepted by J. Phys. Chem. A.June 23, 2025
Anett successfully defends her BSc thesis.June 4, 2025
Kitti successfully defends her MSc dissertation.May 29, 2025
Tibi, Petra, and Balázs give talks at the KeMoMo-QSAR symposium.April 24, 2025
The news about our Nature Communications paper appears on the homepage of the University (see Press)!April 11, 2025
Dorina wins 3rd prize and a special award at the OTDK.April 10, 2025
Dorina, Laura, and Réka give their talks at the OTDK.April 9, 2025
Balázs, Csaba, and Dorina give their talks at the OTDK.March 28, 2025
Balázs successfully defends his PhD dissertation.February 13, 2025
An interview appears on the homepage of MTA apropos of receiving the Lendület grant (see Press)!February 11, 2025
Dóri's paper, written in collaboration with the Wester and Yang groups, on a dynamic isotope effect in the F− + CD3I reaction has been accepted by Nature Communications!February 7, 2025
Timi successfully defends her PhD dissertation.February 6, 2025
Panka Péter (BSc student) joins our group.February 5, 2025
Noémi Szabadszállási (BSc student) joins our group.January 29, 2025
Lili Kószó (BSc student) joins our group.January 16, 2025
Tibi successfully defends his PhD dissertation.January 16, 2025
Gábor's book chapter has been accepted by Advances in Quantum Chemistry.January 8, 2025
Balázs successfully defends his BSc thesis.December 17, 2024
Dóri receives the NKFIH Starting research grant!November 29, 2024
Gábor receives the Momentum Certificate and gives a short talk at the MTA Momentum Day.November 28, 2024
Dorina wins 1st and Balázs wins 2nd place at the Research Student Competition of the University of Szeged.November 22, 2024
Gábor gives a talk in Mátrafüred at the meeting of the MTA Material and Molecular Structure Working Group.November 15, 2024
Domonkos and Erik's F− + CH2ClI benchmark paper has been accepted by J. Phys. Chem. A.November 7, 2024
Petra and Dorina give talks in Mátrafüred at the meeting of the MTA Reaction Kinetics and Photochemistry Working Group.October 31, 2024
Balázs and Attila's paper on the PES and dynamics of the F− + SiH3I reaction has been accepted by J. Chem. Phys.October 31, 2024
András successfully defends his PhD dissertation.October 31, 2024
Laura and Bálint give talks at the KEN conference.October 30, 2024
Our research group is on the Quantum program of Szeged TV (see Press)!October 30, 2024
Dorina and Réka give talks at the KEN conference.October 29, 2024
Petra gives an honorary talk at the KEN conference and receives the Excellence Prize of the Hungarian Chemical Society.October 28, 2024
Balázs's benchmark paper on the OH + CH3NH2 reaction has been accepted by PCCP.October 18, 2024
Petra's MSc dissertation wins an Excellence Prize of the Hungarian Chemical Society.September 24, 2024
Gábor gives an invited talk in Pescara at the Molecular Electronic Structure (MES) conference.September 4, 2024
Petra and Dorina win the scholarship of the University Research Fellowship Program (EKÖP).August 23, 2024
The "Festschrift in honour of Prof. Attila G. Császár", edited by Edit Mátyus and Gábor, has appeared in Molecular Physics.August 12, 2024
Petra, Timi, and Tibi's paper on the PES and dynamics of the Cl + CH3CN reaction has been accepted by J. Chem. Phys.July 15-17, 2024
Roland Wester and four of his group members (Fabio, Christian, Dasarath, and Jerin) visit us from the University of Innsbruck.July 13, 2024
Origo.hu also publishes the Lendület news!July 9, 2024
The news on the new Lendület grant appears on the homepage of the University of Szeged (see Press)!July 2, 2024
Petra successfully passes the entrance exam of the Doctoral School.June 27, 2024
Cangtao's Criegee paper revealing new pathways for the CH2OO + SO2 reaction has been accepted by Communications Chemistry.June 26, 2024
Dóri, Petra, and Viktor present their posters in Istanbul at the Quantum Reactive Scattering (QRS) conference.June 25, 2024
Gábor gives an invited talk in Istanbul at the Quantum Reactive Scattering (QRS) conference.June 18, 2024
Petra receives the gold-level Talent Scholarship of the University of Szeged and the EPAM Scholarship.June 18, 2024
Timi gives a talk about her PhD theses at the Departmental Seminar.June 17, 2024
Csaba successfully defends his BSc thesis.June 14, 2024
Laura, Dorina, Réka, and Bálint successfully defend their BSc theses.June 13, 2024
Petra gives a talk at the online ÚNKP conference of the Institute.June 12, 2024
We have got further Momentum (Lendület, the most prestigious grant in Hungary)!June 3, 2024
Balázs gives a talk about his PhD theses and Domonkos and Dóri give talks on their recent results at the KeMoMo-QSAR symposium.June 1, 2024
Dorina and Dóri's benchmark paper on the Cl + CH3X [X = F, Cl, Br, I] reactions has been accepted by PCCP.May 31, 2024
Petra successfully defends her MSc dissertation.May 30, 2024
Dóri and Timi give talks in Balatonvilágos at the meeting of the MTA Reaction Kinetics and Photochemistry Working Group.May 15-17, 2024
Roland Thissen and David Lauvergnat visit us from the University of Paris-Saclay.May 15, 2024
Kitti, Viktor, and Dóri's paper on the PES and dynamics of the HCl + C2H5 reaction has been accepted by J. Phys. Chem. A.May 10, 2024
Tibi receives a poster prize at the New directions in molecular scattering Faraday Discussion (Edinburgh, United Kingdom).May 10, 2024
Gábor gives an online talk at the New directions in molecular scattering Faraday Discussion (Edinburgh, United Kingdom).May 8, 2024
Balázs and Tibi present their posters at the New directions in molecular scattering Faraday Discussion (Edinburgh, United Kingdom).May 7, 2024
We smash the institutional Scientific Student Competition: Csaba 1st prize, Dorina 1st prize, Laura 1st prize, Réka 2nd prize, and Bálint 3rd prize!May 6, 2024
Domonkos's benchmark paper on the HOO− + CH3Y [Y = F, Cl, Br, I] reactions has been accepted by PCCP.May 3, 2024
András gives a talk about his PhD theses at the Departmental−KeMoMo Seminar.April 16, 2024
Viktor receives the travel support of the Academy.April 4, 2024
Our Perspective article on first-principles mode-specific reaction dynamics, invited to the PCCP 25th Anniversary Collection, has been accepted.March 26, 2024
Petra and Dóri's O− + CD4 paper, written in collaboration with the Wester group, has been accepted by J. Phys. Chem. A.March 5, 2024
Attila's paper on the detailed dynamics of the F− + SiH3Cl reaction has been accepted by PCCP.March 1, 2024
Gábor gives an invited talk at the "Highlighting Organic Chemistry in Hungary" Webinar organized by the Organic Chemistry Division of the European Chemical Society.February 20, 2024
András and Csenge's paper on the PES and dynamics of the OH− + CH3CH2Cl reaction has been accepted by Faraday Discussions.February 12, 2024
Dóri gives an invited talk in the Lennard-Jones Centre Discussion Group series at the University of Cambridge.February 12, 2024
Two BSc students (Márton Felföldi and Anett Major) join the group.January 19, 2024
Timi's F + CH3NH2 JCP paper has been chosen to be promoted as an Editor's Pick.January 18, 2024
Timi's paper on the PES and dynamics of the F + CH3NH2 reaction has been accepted by J. Chem. Phys.January 17, 2024
András receives the CEEPUS Freemover Mobility Scholarship for two months (Innsbruck, Wester group).January 4, 2024
Csenge successfully defends her MSc dissertation.December 25, 2023
Domonkos's mode-specific OH− + CH3I paper has been accepted by J. Chem. Phys.December 14, 2023
In 2025 we will organize the International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) in Szeged!December 12, 2023
Viktor successfully defends his PhD dissertation.December 5, 2023
Anett and Dóri's Chem. Eur. J. paper has been selected by the Chemistry Section of the Hungarian Academy of Sciences as the publication of the month.December 4, 2023
Domonkos successfully defends his PhD dissertation.November 25, 2023
Balázs gives a talk in Mátrafüred at the meeting of the MTA Material and Molecular Structure Working Group.November 24, 2023
Domonkos, András, and Timi give talks in Mátrafüred at the meeting of the MTA Material and Molecular Structure Working Group.November 24, 2023
Petra receives the István Nagypál Scholarship.November 21, 2023
We receive funding from the National Research, Development and Innovation Office for the next 4 years.November 15, 2023
Gábor gives an invited talk at the International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) in Jaipur, India.November 13-15, 2023
Timi and Petra present their posters at the International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) (Shiv Vilas Resorts, Jaipur, India).November 1, 2023
We are in the calendar of the Faculty (see Gallery)!October 19, 2023
Petra gives a talk at the Chemistry Lecture Days.September 29, 2023
Boldi, Timi, and Dóri's benchmark paper on the X− + PH2Y [X, Y = F, Cl, Br, I] reactions has been accepted by PCCP.September 25, 2023
Cangtao's Criegee paper on the PES and dynamics of the CH2OO + NH3 reaction has been accepted by PCCP.September 6, 2023
Timi's paper on the PES and dynamics of the Cl + CH3NH2 reaction has been accepted by J. Chem. Phys.September 5, 2023
Petra wins the scholarship of the New National Excellence Program (ÚNKP).August 8, 2023
Balázs and Viktor's mode-specific OH + C2H6 paper has been accepted by J. Phys. Chem. A.August 8, 2023
Anett and Dóri's paper on the PES and dynamics of the F− + PH2Cl reaction has been accepted by Chem. Eur. J.August 2, 2023
Cangtao's HX (X = Br, I) + C2H5 paper as well as András's F− + CH3CH2I/CF3CH2I paper have been selected for the 2023 HOT PCCP article collection.July 13, 2023
Cangtao's paper on the H/X-abstraction competition in the HX (X = Br, I) + C2H5 reactions has been accepted by PCCP.July 10-11, 2023
András and Balázs present their posters at the Dynamics of Molecular Collisions (DMC) Conference (Snowbird, Utah, USA).July 1, 2023
Petra receives the Excellent Student of the Faculty prize.June 29, 2023
Petra gives a talk at the online ÚNKP conference of the Institute.June 27, 2023
Tibi presents his poster in Bratislava at the 17th International Congress of Quantum Chemistry (ICQC).June 13, 2023
Kitti successfully defends her BSc thesis.June 9, 2023
Attila gives a talk in Balatonvilágos at the meeting of the MTA Reaction Kinetics and Photochemistry Working Group.June 8, 2023
Timi gives a talk in Balatonvilágos at the meeting of the MTA Reaction Kinetics and Photochemistry Working Group.June 8, 2023
Petra receives the silver-level Talent Scholarship of the University of Szeged.June 7, 2023
András's F− + CH3CH2I/CF3CH2I paper, written in collaboration with the Wester and Viggiano groups, has been accepted by PCCP.June 6, 2023
Tibi and Viktor give talks about their PhD theses at the Departmental Seminar.May 24, 2023
Zsolt successfully defends his MSc dissertation.May 18, 2023
Attila's paper on the PES and dynamics of the F− + SiH3Cl reaction has been accepted by J. Chem. Phys.May 18, 2023
Petra, Barnabás, and Cangtao give talks at the KeMoMo-QSAR symposium.May 17, 2023
Domonkos gives a talk about his PhD theses at the Departmental Seminar.April 30, 2023
András and Viktor's paper on the PES and dynamics of the Cl− + CH3I reaction has been accepted by J. Chem. Phys.April 15, 2023
Petra and Máté win 2nd and Anett wins 3rd prize at the OTDK.April 14, 2023
Barnabás, Anett, and Máté give their talks at the OTDK.April 13, 2023
Zsolt, Petra, and Csenge give their talks at the OTDK.March 7, 2023
Cangtao's mode-specific HI + C2H5 paper has been accepted by PCCP.March 6, 2023
András and Máté's paper on the conformers, proton affinities, and gas-phase basicities of serine has been accepted by PCCP.February 1, 2023
Dominika Takó (high-school research student) joins the group.January 17, 2023
Balázs's OH + glycine paper has been accepted by PCCP.January 3, 2023
Csaba Rudner (BSc student) also joins us.January 2, 2023
Four BSc students (Laura Erdei, Dorina Gál, Réka Gyimesi, and Bálint Imre) and three high-school research students (Bartal Baranyi, Péter Fuder, and Nándor Kocsis) join the group.December 20, 2022
Tibi's OH + CH3OH paper has been accepted by J. Chem. Phys.December 16, 2022
Gábor gives a talk in Szentes at the Koszta József Elementary School.December 16, 2022
Domonkos's paper, written in collaboration with the Wester group, on the dynamics of the OH− + CH3I reaction has been accepted by PCCP.December 16, 2022
Boldi successfully defends his BSc thesis.December 15, 2022
Cangtao's mode-specific HBr + C2H5 paper has been accepted by PCCP.December 1, 2022
András's paper on the conformers, proton affinities, and gas-phase basicities of cysteine has been accepted by J. Phys. Chem. A.November 16, 2022
Gábor gives a talk in the MTA-SZAB Hall at the lecture day of the Chemistry Commission celebrating Hungarian Science.November 14, 2022
An interview appears on the homepage of the University of Szeged apropos of the Publication of the Year Prize (see Press)!November 8, 2022
Cangtao's paper on the PES and dynamics of the HI + C2H5 reaction has been accepted by PCCP.November 1, 2022
Cangtao and Viktor's HBr + C2H5 paper has been selected as a 2022 HOT PCCP article.November 1, 2022
Dóri gives an invited talk in Crete at the Stereodynamics conference.October 28, 2022
Balázs gives a talk in Mátrafüred at the meeting of the MTA Reaction Kinetics and Photochemistry Working Group.October 27, 2022
Dóri and András give talks in Mátrafüred at the meeting of the MTA Reaction Kinetics and Photochemistry Working Group.October 22, 2022
András and Tibi give talks in Mátrafüred at the meeting of the MTA Material and Molecular Structure Working Group.October 21, 2022
Gábor gives a talk in Mátrafüred at the meeting of the MTA Material and Molecular Structure Working Group.September 20, 2022
Cangtao and Viktor's paper on the PES and dynamics of the HBr + C2H5 reaction has been accepted by PCCP.September 8, 2022
Domonkos gives a talk in Balatonföldvár at the Quantum Reactive Scattering (QRS) meeting.September 5, 2022
Tibi gives a talk in Balatonföldvár at the Quantum Reactive Scattering (QRS) meeting.September 2, 2022
Petra wins the scholarship of the New National Excellence Program (ÚNKP).September 1, 2022
Gábor receives the Publication of the Year Prize (Nature Chemistry paper) at the University of Szeged.August 8, 2022
Dóri and Viktor's paper, written in collaboration with the Mátyus group (Eötvös University), on the PES and variational vibrational states of CH4.F− has been accepted by Mol. Phys. This is the first spectroscopic application of ROBOSURFER!August 8, 2022
Gábor gives a talk at the departmental seminar of the University of New Mexico.August 2, 2022
Timi's F + CH3NH2 paper has been accepted by PCCP.July 22, 2022
Balázs and Viktor's paper on the PES and dynamics of the OH + C2H6 reaction has been accepted by J. Chem. Phys. This is the first 10-atomic PES in the group!July 13-14, 2022
András, Dóri, Timi, and Viktor present their posters at the Molecular Interactions and Dynamics Gordon Research Conference held in Stonehill College, Easton, USA.July 12, 2022
Tibi gives a talk at the Molecular Interactions and Dynamics Gordon Research Conference held in Stonehill College, Easton, USA.July 11-12, 2022
Tibi presents his poster at the Molecular Interactions and Dynamics Gordon Research Conference held in Stonehill College, Easton, USA.July 11, 2022
Gábor chairs the Molecular Interactions and Collisions in the Gas Phase session at the Molecular Interactions and Dynamics Gordon Research Conference held in Stonehill College, Easton, USA.June 21, 2022
Tibi gives an invited talk at the 3rd YoungCAS Workshop on Global SCF Optimization in Oslo, Norway.June 15, 2022
Petra successfully defends her BSc thesis.June 14, 2022
Anett, Barnabás, and Máté successfully defend their BSc theses.June 9, 2022
András, Balázs, and Timi successfully pass their complex doctoral exams.June 2, 2022
Attila, Domonkos, and Tibi give talks at the KeMoMo-QSAR symposium.May 20, 2022
Petra gives an online talk at the meeting in Balatonvilágos of the MTA Reaction Kinetics and Photochemistry Working Group.May 19, 2022
Petra receives the bronze-level Talent Scholarship of the University of Szeged.May 19, 2022
Gábor and Anett give online talks at the meeting in Balatonvilágos of the MTA Reaction Kinetics and Photochemistry Working Group.May 10, 2022
Domonkos receives the National Young Talent Scholarship.April 27, 2022
Anett wins 1st, Barnabás, Máté, Petra, and Zsolt win 2nd, and Csenge wins 3rd place at the Research Student Competition of the University of Szeged.April 15, 2022
Domonkos's paper on the PES and dynamics of the NH2− + CH3I reaction has been accepted by J. Chem. Phys.April 11, 2022
Petra and Timi's benchmark ab initio Cl + CH3CN paper has been accepted by J. Phys. Chem. A.April 4, 2022
Dóri's rotational mode-specific Cl + C2H6(JK) paper has been accepted by J. Phys. Chem. A.March 24, 2022
The Journal of the Hungarian Chemical Society features our group. Moreover, we are on the front cover (see Press)!March 21, 2022
Viktor's mode-specific F− + CH3CH2Cl paper has been accepted by PCCP.March 7, 2022
Cangtao Yin (postdoc) and Boldizsár Ballay (BSc student) join our group.March 2, 2022
Balázs Molnár (BSc student) joins our group.February 15, 2022
Tibi's ManyHF paper is on the front cover of JCP and selected as Featured!February 9, 2022
Sándor Demes gives a talk at our group seminar.January 31, 2022
Our visitor, Sándor Demes from the Université de Rennes 1 has arrived.January 20, 2022
Tibi's ManyHF paper has been accepted by J. Chem. Phys. as a Communication.January 19, 2022
Zsolt and Domonkos's ambident CN− + CH3Y paper has been accepted by J. Phys. Chem. A.January 12, 2022
Attila successfully passes his complex doctoral exam.November 29, 2021
Our Nature Chemistry paper has been selected by the Chemistry Section of the Hungarian Academy of Sciences as the publication of the month.November 27, 2021
Petra gives a talk at Móra Interdisciplinary Conference.November 22, 2021
Petra gives a talk at Móra Open University.November 13, 2021
Gábor receives the Young Researcher of the Year certificate of merit.November 10, 2021
Domonkos's oxide ion substitution paper is on the back cover of Chemical Science and has been selected for the 2021 Chemical Science HOT Article Collection!November 10, 2021
Domonkos receives the Scholarship for the Young Talents of the Nation.October 22, 2021
Balázs, Domonkos, and Timi give talks in Mátrafüred at the meeting of the MTA Reaction Kinetics and Photochemistry Working Group.October 21, 2021
Dóri gives a talk in Mátrafüred at the meeting of the MTA Reaction Kinetics and Photochemistry Working Group.October 13, 2021
Attila and Gyula's paper on the C- vs. Si-centered SN2 reactions has been accepted by J. Phys. Chem. A.October 13, 2021
Domonkos's paper on the discovery of a new oxide ion substitution pathway for the OH− + CH3F reaction has been accepted by Chem. Sci.October 8, 2021
Csenge receives the Zoltán Bay scholarship from the Council of Gyula City.October 6, 2021
Gábor gives a talk at the University of Pécs.September 30, 2021
Dóri, Viktor, and András give talks at the KeMoMo-QSAR symposium.September 28, 2021
Gábor gives an online invited talk at the "Machine Learning and Informatics for Chemistry and Materials" Telluride Workshop.September 27, 2021
Attila's paper on the proton affinity of alanine has been accepted by J. Comput. Chem.September 24, 2021
Dóri's paper on the mode-specific dynamics of the F + C2H6 reaction has been accepted by J. Chem. Phys.September 1, 2021
Kitti Horváth (BSc student) joins our group.August 30, 2021
Viktor and Tibi's paper on the dynamics of the F− + CH3Br reaction has been accepted by J. Chem. Phys.August 28, 2021
Dóri's paper on the mode-specific dynamics of the Cl + C2H6 reaction has been accepted by J. Chem. Phys.August 12, 2021
The homepage of the Hungarian Academy of Sciences features our Nature Chemistry article (see Press)!August 10, 2021
The news about our Nature Chemistry paper appears on the homepage of the University and on the delmagyar.hu (see Press)!August 9, 2021
Our Nature Chemistry paper has been published online!August 2, 2021
The Momentum groups of our department appear on the delmagyar.hu (see Press).July 27, 2021
The Momentum groups of our department are showcased on the homepage of the University (see Press).July 6, 2021
Domonkos and Csenge's OH− + CH3CH2Y paper has been selected as a 2021 HOT PCCP article.June 16, 2021
Csenge successfully defends her BSc thesis.June 15, 2021
Viktor and Tibi's paper has been accepted by Nature Chemistry!June 15, 2021
Zsolt successfully defends his BSc thesis.May 31, 2021
Demeter successfully defends his MSc dissertation.May 27, 2021
Domonkos, Tibi, and Attila give talks at the online meeting of the MTA Reaction Kinetics and Photochemistry Working Group.May 24, 2021
Domonkos and Csenge's OH− + CH3CH2Y paper has been accepted by PCCP.May 18, 2021
Balázs gives a talk at the Student Research (OTDK) Conference.April 28, 2021
András's glycine paper is on the cover of PCCP!April 21, 2021
Dóri's multiple-inversion paper is on the cover of Chemical Science!April 13, 2021
Gábor gives a talk at the Bill Hase Memory Symposium of the 2021 Spring ACS Meeting.April 2, 2021
Timi's Cl + CH3NH2 paper has been accepted by PCCP.April 1, 2021
Our Perspective is on the cover of JPCA and has been among the Most Read articles!March 29, 2021
András's paper on the proton affinity of glycine has been accepted by PCCP.March 12, 2021
Dóri's paper revealing a multiple inversion mechanism for F− + NH2Cl has been accepted by Chem. Sci.February 22, 2021
Gábor's portrait appears on the homepage of MTA showcasing the most successful researchers supported by the Bolyai Scholarship of the Hungarian Academy of Sciences (see Press).February 16, 2021
Our Perspective article has been accepted by J. Phys. Chem. A.February 3, 2021
Máté Kígyósi (BSc student) joins our group.January 4, 2021
Gábor gives an online invited talk at the Beijing Institute of Technology.November 11, 2020
Viktor successfully passes his complex doctoral exam.November 6, 2020
Viktor gives a talk at the online meeting of the MTA Reaction Kinetics and Photochemistry Working Group.October 2, 2020
Paszkál's paper on the JK-dependent dynamics of the F− + CH3I(JK) reaction has been accepted by J. Phys. Chem. A.September 23, 2020
Balázs wins 1st place at the Research Student Competition of the University of Szeged.September 15, 2020
Tibi's ROBOSURFER article becomes a "Highly Cited Paper" (top 1% of its academic field) on Web of Science.September 1, 2020
Balázs, András, and Timi begin their PhD work, and three BSc students (Anett Giricz, Barnabás Sipos, and Petra Tóth) join the group.August 28, 2020
Timi successfully passes the entrance exam of the Doctoral School.July 19, 2020
Dóri's paper on the PES and dynamics of the F + C2H6 reaction has been accepted by J. Chem. Phys.July 7, 2020
Paszkál and Viktor's paper on the separation of the front-side attack and double-inversion SN2 pathways has been accepted by Chem. Phys. Lett.June 26, 2020
András and Balázs successfully pass the entrance exam of the Doctoral School.June 22, 2020
Balázs's OH + CH4/C2H6 paper has been accepted by PCCP.June 10, 2020
Erik and András's paper on the conformers of dehydrogenated glycine isomers has been accepted by J. Comput. Chem.June 1, 2020
Tímea Szűcs joins our group.May 28, 2020
Balázs and Erik successfully defend their MSc dissertations.May 27, 2020
Tibi successfully passes his complex doctoral exam.May 22, 2020
Dóri, Viktor, and Tibi's paper on the PES and dynamics of the Cl + C2H6 reaction has been accepted by J. Phys. Chem. Lett.May 22, 2020
Anna successfully defends her BSc thesis.April 6, 2020
Two of our papers (OH− + CH3I and CH4.Ar PESs) have been selected as 2020 HOT PCCP articles.February 26, 2020
Our Perspective article is on the front cover of PCCP!February 21, 2020
Our renewed offices are ready!February 3, 2020
Tibi's ROBOSURFER paper is the most read article in JCTC for the previous 30 days. On January 16 it was 5th and on January 27 it was 4th among the most read articles.February 3, 2020
Attila Dékány (PhD student), Paszkál Papp (MSc project student), and Adrián Traj (MSc project student) join our group.January 27, 2020
Domonkos and Tibi's paper on the PES of the OH− + CH3I reaction has been accepted by PCCP as a Communication.January 14, 2020
Dorina Szepesi (high-school research student) joins our group.December 20, 2019
Dóri's CH4.Ar paper, written in collaboration with the Mátyus group (Eötvös University), has been accepted by PCCP.December 18, 2019
Tibi's ROBOSURFER paper has been accepted by JCTC.December 6, 2019
Balázs successfully defends his PhD dissertation.December 6, 2019
Our Perspective article has been accepted by PCCP.November 8, 2019
Gábor gives a talk in Mátrafüred at the meeting of the MTA Reaction Kinetics and Photochemistry Working Group.November 7, 2019
Domonkos gives a talk in Mátrafüred at the meeting of the MTA Reaction Kinetics and Photochemistry Working Group.November 6, 2019
Dóri gives a talk in the MTA-SZAB Hall at the "Young chemists serving science" lecture day.October 30, 2019
Domonkos gives a talk at the Chemistry Lecture Days.September 19, 2019
Gábor gives a talk at the seminar of the Department of Theoretical Physics.September 18, 2019
Gábor is an invited speaker at the Eötvös Chemistry Workshop.September 5, 2019
Our research group is introduced on the Quantum program of Szeged TV.July 18, 2019
Our group hosts a Momentum grant party in our Department.July 4, 2019
Gábor gives an invited talk in Tokyo at the Quantum Reactive Scattering (QRS) meeting.July 3, 2019
Dóri presents her poster in Tokyo at the Quantum Reactive Scattering (QRS) meeting.July 1, 2019
Gábor becomes Associate Professor.June 27, 2019
Gábor receives the prestigious Momentum grant.June 22, 2019
Five members of our group (Gábor, Dóri, Domonkos, Tibi, and Viktor) arrive in Edinburgh to participate at the International Symposium on Molecular Beams (ISMB).June 6, 2019
Gábor gives a talk at the KeMoMo-QSAR symposium.May 30, 2019
Csenge Tokaji (BSc student) joins our group.May 30, 2019
Domonkos successfully passes his complex doctoral exam.May 27, 2019
Demeter successfully defends his BSc thesis.May 15, 2019
Zsolt Kerekes (BSc student) joins our group.May 14, 2019
Balázs gives a successful PhD pre-defense talk.May 9, 2019
Domonkos receives certificate of the Talent scholarship in the PhD category.May 6, 2019
Balázs successfully passes his final doctoral exam.April 4, 2019
Domonkos gives a talk in Debrecen at the I. FKF symposium.March 15, 2019
Domonkos and Zita's second paper on SN2 reactions has been accepted by PCCP.February 4, 2019
Anna Schmidt (BSc student) joins our group.February 1, 2019
Viktor begins his PhD work.December 20, 2018
At 20:10 Balázs's JPCA paper is accepted. At 22:33 Balázs's PCCP paper is accepted.December 20, 2018
Domonkos receives the National Young Talent Scholarship.December 14, 2018
Viktor successfully defends his MSc dissertation.December 4, 2018
Gábor's paper on the dynamics of proton transfer from ArH+ to CO, written in collaboration with the Wester group, has been accepted by Int. J. Mass Spectrom.November 30, 2018
Dóri and Balázs Gruber's paper on the X + C2H6 [X = F, Cl, Br, I] reactions has been accepted by Phys. Chem. Chem. Phys.November 22, 2018
Viktor wins 2nd place at the Research Student Competition of the University of Szeged.November 8, 2018
Dóri, Domonkos, Viktor, Tibi, and Balázs give talks in Veszprém at the meeting of MTA Reaction Kinetics and Photochemistry Working Group.October 16, 2018
Balázs and Domonkos give talks at the KEN conference.October 15, 2018
Tibi wins an Excellence Prize of the Hungarian Chemical Society and gives an honorary talk at the KEN conference.October 1, 2018
Balázs wins a 6-months Richter scholarship.September 19, 2018
Balázs's paper on the mode-specific dynamics of the F− + CH3I reaction has been accepted by J. Phys. Chem. A.July 7, 2018
Gábor receives the Science Prize of the Faculty of Science and Informatics.June 29, 2018
Gábor receives his certificate of habilitation at the University of Szeged.June 28, 2018
Gábor receives the Bolyai Plaquette from the president of the Hungarian Academy of Sciences.June 7, 2018
Domonkos and Zita's paper on the OH− + CH3Y [Y = F, Cl, Br, I] SN2 reactions has been accepted by J. Phys. Chem. A.June 4, 2018
Tibi successfully defends his MSc dissertation.May 29, 2018
András, Erik, and Balázs successfully defend their BSc theses.May 24, 2018
Balázs, Domonkos, Tibi, and Viktor give talks at the KeMoMo-QSAR 2018 Symposium.May 9, 2018
Balázs's paper prepared in an international collaboration gets accepted in Science Advances.May 9, 2018
Gábor gives a talk at the "Quantum Chemistry in Hungary" meeting of the Hungarian Academy of Sciences.April 18, 2018
Gábor gives an invited talk at the Anharmonicity in medium-sized molecules and clusters (AMOC) Meeting in Budapest.March 16, 2018
Tibi's first paper appears in J. Phys. Chem. A reporting an unprecedented investigation on the effects of the electronic structure theory on the dynamics of a chemical reaction.February 27, 2018
Gábor gives a successful habilitation lecture.January 23, 2018
Bálint’s paper on N-centered SN2 reactions appears in J. Phys. Chem. A.January 1, 2018
Dóra Papp joins our group as a postdoctoral researcher.December 12, 2017
Gábor gives an invited talk at a COST Workshop in Ciudad Real, Spain.December 6, 2017
Gábor obtains his DSc degree and becomes the youngest doctor of the Hungarian Academy of Sciences.November 30, 2017
Our invited Feature Article on SN2 reactions appears on the front cover of J. Phys. Chem. A.November 21, 2017
Laci’s paper appears in J. Phys. Chem. A revealing novel mechanisms for the Cl + CH4 -> H + CH3Cl reaction.October 29, 2017
Gábor gives an invited talk at an SN2 Reaction Dynamics Symposium held at an ACS Southwest Regional Meeting in Lubbock, Texas, USA.September 12, 2017
Gábor’s new Nature Chemistry paper on the angle-dependent barrier of the Cl + CHD3 reaction appears in collaboration with the leading experimental group of Kopin Liu.September 1, 2017
Domonkos Attila Tasi (PhD student), Balázs Gruber (BSc student), and Erik Orján (BSc student) join our group.July 13, 2017
Gábor receives a four-year grant from the National Research, Development and Innovation Office (NKFIH).July 10, 2017
István’s paper on the Cl− + CH3I reaction appears in J. Phys. Chem. A.July 3, 2017
Gábor gives an invited talk at the XIV International Workshop on Quantum Reactive Scattering in Trieste, Italy.June 29, 2017
Zita gives a successful talk about her MSc project work.June 20, 2017
Gábor gives an invited talk at the 8th International Meeting on Atomic and Molecular Physics and Chemistry (IMAMPC) in Torun, Poland.June 9, 2017
The TOP paper of István and Balázs appears in J. Phys. Chem. Lett. deciphering front-side complex formation in SN2 reactions via trajectory orthogonal projection (TOP).May 26, 2017
Gyula defends his BSc dissertation.March 24, 2017
Viktor’s paper on the best ab initio study for the F− + CH3CH2Cl reaction appears in J. Phys. Chem. A.March 7, 2017
Demeter Ván (BSc student) joins our group.February 15, 2017
Balázs’s first paper on the potential energy surface and dynamics of the F− + CH3I reaction has been accepted in Chemical Science.February 1, 2017
László Krotos (MSc student) joins our group.January 26, 2017
Bálint gives a successful talk about his MSc project work.January 25, 2017
Gábor defends his Doctor of the Hungarian Academy of Sciences (DSc) dissertation.December 22, 2016
Viktor defends his BSc dissertation.November 24, 2016
Tibi wins 1st place at the Research Student Competition of the University of Szeged.October 3, 2016
István’s paper on the multi-channel mode-specific dynamics of the F− + CHD2Cl reaction appears in J. Chem. Phys.September 1, 2016
Many new students, Zita Fábián (MSc), Bálint Hajdu (MSc), Gyula Kovács (BSc), András Nacsa (BSc), and Viktor Tajti (BSc), join our group.August 9, 2016
Our paper on the highest-dimensional quantum dynamics on a SN2 reaction appears in J. Phys. Chem. Lett. in collaboration with Professors Minghui Yang and Hua Guo.July 12, 2016
István defends his PhD dissertation entitled "Dynamics of biomolecular nucleophilic substitution and proton-transfer reactions on global ab initio potential energy surfaces". He continues his work in the King’s College London, as a postdoctoral associate in the group of Professor Rosta.May 24, 2016
Tibi defends his BSc dissertation and Gergő gives a successful talk about his MSc project work.February 1, 2016
Gergő Szekeres joins us as a MSc project student.November 30, 2015
Our paper appears in Nature Chemistry on the unexpected leaving group effect in SN2 reactions in collaboration with the Wester group in Innsbruck.November 17, 2015
Our paper [Nature Communications 6, 5972 (2015)], which was previously highlighted by the National Geographic Hungary, Index.hu, and the Hungarian Television, on the new double-inversion mechanism becomes a "Highly Cited Paper" on Web of Science.September 7, 2015
Gábor gives an invited talk at the 14th Central European Symposium on Theoretical Chemistry (CESTC) in Banská Bystrica, Slovakia.September 1, 2015
István Szabó (research assistant), Balázs Olasz (PhD student), and Tibor Győri (BSc student) join the group.August 1, 2015
Gábor Czakó joins the Faculty of the University of Szeged as Assistant Professor and becomes the head of the new Computational Reaction Dynamics Research Group.Media appearances
Science: art! – The SZTE Doctoral School of Chemistry commemorative medal was awarded for the first time (in Hungarian)
On the homepage of SZTE, apropos of the first medal ceremony at Attila Dékány's PhD defense (2025)
Dr. Dóra Papp and Dr. Gábor Czakó described the quantum tunneling effect of a chemical reaction in a Nature Communications article (in Hungarian)
On the homepage of SZTE, apropos of the appearance of our Nature Communications article (2025)
Featured Lendület Researcher: Gábor Czakó
On the homepage of MTA, apropos of receiving another Lendület grant (2025)
Our group on Szeged Television (in Hungarian)
Szeged TV Quantum (from 7:43), apropos of receiving another Lendület grant (2024)
Quantum effects on the atomic terrain table - Dr. Gábor Czakó won his second MTA Lendület grant (in Hungarian)
On the homepage of SZTE, apropos of receiving another Lendület grant (2024)
Dr. Gábor Czakó (University of Szeged) associate professor: "Computational chemistry can be more accurate than experiment" (in Hungarian)
On the homepage of SZTE, apropos of the Publication of the Year Prize (2022)
Theoretical reaction dynamics research at the University of Szeged (in Hungarian)
About our research group in the Journal of the Hungarian Chemical Society (Magyar Kémikusok Lapja) (2022) Cover
Momentum researchers first identify the experimental fingerprint of a chemical reaction (in Hungarian)
On the homepage of MTA, apropos of the appearance of our 3rd Nature Chemistry article (2021)
Researchers at Szeged publish an article about the chemical reaction of a complex system (in Hungarian)
On Delmagyar.hu, apropos of the appearance of our 3rd Nature Chemistry article (2021)
Chemistry without chemicals – New Nature Chemistry article from the SZTE Czakó group (in Hungarian)
On the homepage of SZTE, apropos of the appearance of our 3rd Nature Chemistry article (2021)
Momentum: six researchers won support in Szeged (in Hungarian)
On Delmagyar.hu, introduction of the Momentum groups of our Department (2021)
The Department of Physical Chemistry and Materials Science at the University of Szeged is the most "Momentous" (in Hungarian)
On the homepage of SZTE, introduction of the Momentum groups of our Department (2021)
"Bolyaisok" – Gábor Czakó theoretical chemist (in Hungarian)
On the homepage of MTA, showcasing the most successful researchers supported by the Bolyai Scholarship of the Hungarian Academy of Sciences (2021)
University of Szeged gives prizes to its excellent teachers and researchers (in Hungarian)
On the homepage of SZTE, apropos of Gábor receiving the Young Researcher of the Year certificate of merit (2020)
Our group on Szeged Television (in Hungarian)
Szeged TV Quantum (from 7:15), apropos of receiving the Lendület grant (2019)
Energy efficiency research in Szeged: Group of Gábor Czakó may join the international frontline (in Hungarian)
In Délmagyarország, apropos of receiving the Lendület grant (2019)
Gábor Czakó receives Momentum research grant (in English)
On the homepage of Emory University, apropos of receiving the Momentum (Lendület) grant (2019)
Two researchers of the University of Szeged, Gábor Czakó and Éva Anna Enyedy won at the MTA Lendület program in 2019 (in Hungarian)
On the homepage of SZTE, apropos of receiving the Lendület grant (2019)
From the medieval music records to the risk prediction of the coronary artery disease – the new winners of the MTA Lendület program (in Hungarian)
On the homepage of the Hungarian Academy of Sciences, apropos of receiving the Lendület grant (2019)
Collaboration of theory and experiment (in Hungarian)
Interview about our research in the Journal of the Hungarian Chemical Society (Magyar Kémikusok Lapja) (2019)
Publishing and programming (in Hungarian)
In CAMPUS Plusz (eduline), about our research group in a HVG magazine (2018)
Breaking the bond: To take part or not? (in English)
On ScienceDaily, apropos of the appearance of a Science Advances article (2018)
Theoretical chemistry and non-linear dynamics research in the Institute of Chemistry of the University of Szeged (in Hungarian)
About our research group in the Journal of the Hungarian Chemical Society (Magyar Kémikusok Lapja) (2018)
Gábor Czakó, theoretical chemist: "Unpublished results of a researcher simply do not exist" (in Hungarian)
On the homepage of SZTE, apropos of the appearance of a new Nature Chemistry article (2017)
People read about SZTE in the USA! (in Hungarian)
On SZEGEDMA online, apropos of the appearance of a Feature Article in J. Phys. Chem. A (2017)
Study clarifies the role of the leaving group in gas-phase bimolecular reaction (in English)
On phys.org, apropos of the appearance of the group's first Nature Chemistry article (2015)
Chemists from the University of Szeged in the world-leading chemistry journal (in Hungarian)
On SZEGEDMA online, apropos of the appearance of the group's first Nature Chemistry article (2015)
Groundbreaking discovery of chemists from the University of Szeged (in Hungarian)
In Délmagyarország, apropos of the appearance of the group's first Nature Chemistry article (2015)
Achievements of chemists from the University of Szeged highlighted in the world-leading chemistry journal (in Hungarian)
On the homepage of SZTE, apropos of the appearance of the group's first Nature Chemistry article (2015)
Gallery