A kutatócsoportról dióhéjban
|
Jelenlegi munkatársak: 20 Cikkek: 97 Nature Chemistry cikkek: 3 PhD disszertációk: 9 MSc diplomamunkák: 9 BSc szakdolgozatok: 24 MSc projektmunkák: 6 TDK dolgozatok: 16 |

|
Csoporttagok
Csoportvezető
Prof. Dr. Czakó GáborEgyetemi tanár, az MTA doktora
Fizikai Kémiai és Anyagtudományi Tanszék, Szegedi Tudományegyetem
Rerrich Béla tér 1, 6720 Szeged, Magyarország
Iroda: BE-417, Tel.: +36-62-34-3742
E-mail: gczako at chem.u-szeged.hu
Külső munkatársak
 Dr. Papp Dóra
Dr. Papp DóraEgyetemi adjunktus
Posztdoktor a csoportban (2018-2024)
Iroda: BE-420
E-mail: dorapapp at chem.u-szeged.hu
Honlap
Posztdoktori kutatók

Dr. Dékány Attila
Posztdoktor (2025 -), Doktorjelölt (2024-2025), PhD hallgató (2020-2024)
Iroda: BE-418
E-mail: dekanyattilaadam at gmail.com

Dr. Gruber Balázs
Posztdoktor (2025 -), Doktorjelölt (2024-2025), PhD hallgató (2020-2024)
MSc hallgató (2018-2020), BSc hallgató (2017-2018)
E-mail: gbalazs96 at gmail.com

Dr. Győri Tibor
Posztdoktor (2025 -), Doktorjelölt (2022-2025), PhD hallgató (2018-2022)
MSc hallgató (2016-2018), BSc hallgató (2015-2016)
Iroda: BE-414
E-mail: tiborgyri at gmail.com

Dr. Tajti Viktor
Posztdoktor (2024 -), Doktorjelölt (2023-2024), PhD hallgató (2019-2023)
MSc hallgató (2017-2019), BSc hallgató (2016-2017)
Iroda: BE-414
E-mail: vtajti at chem.u-szeged.hu
PhD hallgatók

Tóth Petra
PhD hallgató (2024 - ), MSc hallgató (2022-2024), BSc hallgató (2020-2022)
Iroda: BE-414
E-mail: toth.petra.294 at gmail.com
Kutató munkatársak

Dr. Nacsa András
Kutató munkatárs (2024 - ), Doktorjelölt (2024), PhD hallgató (2020-2024)
Kutató munkatárs (2018-2020), BSc hallgató (2016-2018)
E-mail: nacsa.andras.bence at gmail.com

Dr. Szűcs Tímea
Kutató munkatárs (2025 - ), Doktorjelölt (2024-2025), PhD hallgató (2020-2024), Kutató munkatárs (2020)
E-mail: szucsmeja at gmail.com
MSc hallgatók

Imre Bálint
MSc hallgató (2024 - ), BSc hallgató (2023-2024)
Iroda: BE-418
E-mail: balintimre0902 at gmail.com

Molnár Balázs
MSc hallgató (2025 - ), BSc hallgató (2022-2025)
E-mail: 2017molbal at gmail.com

Rudner Csaba
MSc hallgató (2024 - ), BSc hallgató (2023-2024)
Iroda: BE-418
E-mail: rudnercsaba02 at gmail.com

Sárik Levente
(2025 - )
E-mail: levi200320 at gmail.com
BSc hallgatók

Bohr Anna
(2025 - )
E-mail: bohrpanka at gmail.com

Felföldi Márton
(2024 - )
E-mail: bestfmc5 at gmail.com

Jancsó István
(2025 - )
E-mail: jisti88 at gmail.com

Kószó Lili
(2025 - )
E-mail: koszolili10 at gmail.com

Péter Panka Viola
(2025 - )
E-mail: peterpanka.school at gmail.com

Szabadszállási Noémi
(2025 - )
E-mail: szabadszallasinoemi at gmail.com

Takó Dominika
BSc hallgató (2025 - ), Középiskolás kutatódiák (2023-2025)
E-mail: tako.dominika at gmail.com
Korábbi csoporttagok

Ballay Boldizsár
BSc hallgató (2022-2023)

Baranyi Bartal
Középiskolás kutatódiák (2023)

Erdei Laura
MSc hallgató, projektmunka (2024-2025), BSc hallgató (2023-2024)

Fábián Zita
MSc hallgató, projektmunka (2017)

Fuder Péter
Középiskolás kutatódiák (2023)

Gál Dorina
Kutató munkatárs (2024-2025), BSc hallgató (2023-2024)
E-mail: dorinagalgd at gmail.com

Giricz Anett
BSc hallgató (2020-2022)

Gyimesi Réka
Kutató munkatárs (2024-2025), BSc hallgató (2023-2024)
E-mail: mesireka at gmail.com

Hajdu Bálint
MSc hallgató, projektmunka (2016)

Horváth Kitti
MSc hallgató (2023-2025), BSc hallgató (2021-2023)

Kerekes Zsolt
MSc hallgató (2021-2023), BSc hallgató (2019-2021)

Kígyósi Máté
BSc hallgató (2021-2022)

Kocsis Nándor
Középiskolás kutatódiák (2023)

Kovács Gyula
BSc hallgató (2016-2017)

Krotos László
MSc hallgató, projektmunka (2017)

Major Anett
BSc hallgató (2024-2025)

Dr. Olasz Balázs
PhD hallgató (2015-2019)

Orján Erik
MSc hallgató (2018-2020), BSc hallgató (2017-2018)

Papp Paszkál
MSc hallgató, projektmunka (2020)

Schmidt Anna Katinka
BSc hallgató (2019-2020)

Sipos Barnabás
Kutató munkatárs (2022-2023), BSc hallgató (2020-2022)

Dr. Szabó István
PhD hallgató (2012-2016)

Szekeres Gergő Péter
MSc hallgató, projektmunka (2016)

Szepesi Dorina
Középiskolás kutatódiák (2020)

Dr. Tasi Domonkos Attila
Posztdoktor (2023-2024), Doktorjelölt (2021-2023), PhD hallgató (2017-2021)
Honlap

Tokaji Csenge
MSc hallgató (2021-2024), BSc hallgató (2019-2021)

Traj Adrián
MSc hallgató, projektmunka (2020)

Ván Demeter
MSc hallgató (2019-2021), BSc hallgató (2017-2019)

Dr. Cangtao Yin
Posztdoktor (2022-2024)
Kutatás
Benchmark ab initio termokémia
Modern elektronszerkezet-számító módszereket alkalmazunk reaktív kémia rendszerekhez tartozó stacionárius pont tulajdonságok, úgymint szerkezetek, relatív energiák, harmonikus rezgési frekvenciák lehető legpontosabb meghatározására. A számított eredmények benchmark termokémiai adatokat szolgáltatnak és segítik a kísérleti és elméleti munkák tervezését. Továbbá, a stacionárius pontok feltérképezése az első lépés kémiai reakciók potenciálisenergia-felületeinek fejlesztése és dinamikai vizsgálata felé.
Potenciálisenergia-felületek fejlesztése
A potenciálisenergia-felületek (PES) irányítják az atomok mozgását egy kémiai reakció során. A csoportunkban pontos ab initio energiapontok illesztésével analitikus globális PES-eket fejlesztünk reaktív kémiai rendszerekre. PES-eink minden korábbinál részletesebb és pontosabb dinamikai vizsgálatokat tesznek lehetővé. Az X + CH4 [X = F, O, Cl, Br] és a F− + CH3Y [Y = F, Cl, I] reakciókra már vannak pontos teljes-dimenziós PES-eink és jelenleg is fejlesztünk PES-eket számos érdekes kémiai rendszerre egészen 12 atomig.
Reakciódinamika számítások
Az analitikus PES-ek lehetővé teszik a reakciódinamika tanulmányozását a kvázi-klasszikus trajektória és/vagy – együttműködésben vezető kínai és amerikai csoportokkal – kvantummechanikai módszerek alkalmazásával. A dinamika szimulációk lépésről-lépésre követik az atomok mozgását, így új reakcióutakat tárhatnak fel és a reakciók kimenetele is jósolhatóvá válik. Számításaink segíthetik és motiválhatják a kísérleti vizsgálatokat, ezért is vannak aktív együttműködéseink vezető osztrák és tajvani kísérleti csoportokkal.
Alkánok reakciódinamikája atomokkal és gyökökkel
Tanulmányoztuk a metán reakciódinamikáját F, O, Cl és Br atomokkal, amely munkák sok jelentős felfedezést hoztak, úgymint a Nobel-díjas John Polanyi szabályainak módosítása poliatomos rendszerek esetén, rezgési és forgási mód specificitás, szögfüggő energiagát, új reakcióutak, stb. Eredményeinket a világ legjobb tudományos folyóiratai, úgymint a Science, Nature Chemistry, PNAS és JACS közölték.
SN2 reakciók dinamikája
A bimolekuláris nukleofil szubsztitúciós (SN2) reakciók - melyek Walden-inverziós mechanizmusa már több mint száz éve ismert - központi szerepet játszanak a kémiában és a biokémiában. Az analitikus PES-ek alkalmazásával nemrégiben egy új kutatási irányt indítottunk az SN2 reakciók dinamikájának területén. Szimulációink feltártak (a) egy új SN2 mechanizmust, ami dupla inverzió nevet kapta, (b) meglepő távozócsoport hatást, (c) elölről-támadásos komplex képződésén alapuló nem-tradicionális dinamikát, (d) minden korábbinál jobb egyezést a kísérlettel, stb. A fenti eredmények magas impakt faktorú folyóiratokban - Nature Communications, Nature Chemistry és Chemical Science – kerültek publikálásra, felkeltették a hazai és nemzetközi média érdeklődését és a Journal of Physical Chemistry A folyóirat a címlapján emelte ki őket.
Kis biorendszerek dinamikája
Jelenleg új PES reprezentációs módszerek fejlesztésén dolgozunk, amelyek lehetővé teszik kis biorendszerek tanulmányozását, úgymint aminosavak konformációs dinamikájának, valamint atomokkal és gyökökkel végbemenő reakcióinak vizsgálatát. Szimulációink minden bizonnyal pontosabbak és megbízhatóbbak lesznek, mint a klasszikus erőtereken alapuló korábbi irodalmi eredmények.
Publikációk 2015. augusztus óta
I. Szabó and G. Czakó
Rotational mode specificity in the F− + CH3Y [Y = F and Cl] SN2 reactions
J. Phys. Chem. A 119, 12231 (2015) PDF
M. Stei, E. Carrascosa, M. A. Kainz, A. H. Kelkar, J. Meyer, I. Szabó, G. Czakó, and R. Wester
Influence of the leaving group on the dynamics of a gas-phase SN2 reaction
Nat. Chem. 8, 151 (2016) PDF Highly Cited Paper (as of July/August 2016)
Y. Wang, H. Song, I. Szabó, G. Czakó, H. Guo, and M. Yang
Mode-specific SN2 reaction dynamics
J. Phys. Chem. Lett. 7, 3322 (2016) PDF
I. Szabó and G. Czakó
Mode-specific multi-channel dynamics of the F− + CHD2Cl reaction on a global ab initio potential energy surface
J. Chem. Phys. 145, 134303 (2016) PDF
B. Olasz, I. Szabó, and G. Czakó
High-level ab initio potential energy surface and dynamics of the F− + CH3I SN2 and proton-transfer reactions
Chem. Sci. 8, 3164 (2017) PDF
V. Tajti and G. Czakó
Benchmark ab initio characterization of the complex potential energy surface of the F− + CH3CH2Cl reaction
J. Phys. Chem. A 121, 2847 (2017) PDF
I. Szabó, B. Olasz, and G. Czakó
Deciphering front-side complex formation in SN2 reactions via dynamics mapping
J. Phys. Chem. Lett. 8, 2917 (2017) PDF
I. Szabó and G. Czakó
Benchmark ab initio characterization of the complex potential energy surface of the Cl− + CH3I reaction
J. Phys. Chem. A 121, 5748 (2017) PDF
H. Pan, F. Wang, G. Czakó, and K. Liu
Direct mapping of the angle-dependent barrier to reaction for Cl + CHD3 using polarized scattering data
Nat. Chem. 9, 1175 (2017) PDF
I. Szabó and G. Czakó
Dynamics and novel mechanisms of SN2 reactions on ab initio analytical potential energy surfaces
J. Phys. Chem. A 121, 9005 (2017) Feature Article Cover PDF
L. Krotos and G. Czakó
Does the Cl + CH4 __> H + CH3Cl reaction proceed via Walden inversion?
J. Phys. Chem. A 121, 9415 (2017) PDF
B. Hajdu and G. Czakó
Benchmark ab initio characterization of the complex potential energy surfaces of the X− + NH2Y [X,Y = F, Cl, Br, I] reactions
J. Phys. Chem. A 122, 1886 (2018) PDF
T. Győri, B. Olasz, G. Paragi, and G. Czakó
Effects of the level of electronic structure theory on the dynamics of the F− + CH3I reaction
J. Phys. Chem. A 122, 3353 (2018) PDF
S. Góger, P. Szabó, G. Czakó, and G. Lendvay
Flame inhibition chemistry: rate coefficients of the reactions of HBr with CH3 and OH radicals at high temperatures determined by quasiclassical trajectory calculations
Energy Fuels 32, 10100 (2018) PDF
M. Stei, E. Carrascosa, A. Dörfler, J. Meyer, B. Olasz, G. Czakó, A. Li, H. Guo, and R. Wester
Stretching vibration is spectator in nucleophilic substitution
Sci. Adv. 4, eaas9544 (2018) PDF
D. A. Tasi, Z. Fábián, and G. Czakó
Benchmark ab initio characterization of the inversion and retention pathways of the OH− + CH3Y [Y = F, Cl, Br, I] SN2 reactions
J. Phys. Chem. A 122, 5773 (2018) PDF
B. Olasz and G. Czakó
Mode-specific quasiclassical dynamics of the F− + CH3I SN2 and proton-transfer reactions
J. Phys. Chem. A 122, 8143 (2018) PDF
D. Papp, B. Gruber, and G. Czakó
Detailed benchmark ab initio mapping of the potential energy surfaces of the X + C2H6 [X = F, Cl, Br, I] reactions
Phys. Chem. Chem. Phys. 21, 396 (2019) PDF
B. Bastian, E. Carrascosa, A. Kaiser, J. Meyer, T. Michaelsen, G. Czakó, W. L. Hase, and R. Wester
Dynamics of proton transfer from ArH+ to CO
Int. J. Mass Spectrom. 438, 175 (2019) PDF
B. Olasz and G. Czakó
High-level-optimized stationary points for the F−(H2O) + CH3I system: Proposing a new water-induced double-inversion pathway
J. Phys. Chem. A 123, 454 (2019) PDF
B. Olasz and G. Czakó
Uncovering the role of the stationary points in the dynamics of the F− + CH3I reaction
Phys. Chem. Chem. Phys. 21, 1578 (2019) PDF
D. A. Tasi, Z. Fábián, and G. Czakó
Rethinking the X− + CH3Y [X = OH, SH, CN, NH2, PH2; Y = F, Cl, Br, I] SN2 reactions
Phys. Chem. Chem. Phys. 21, 7924 (2019) PDF
G. Czakó
Alapvetõ kémiai reakciók dinamikájának és mechanizmusainak vizsgálata
Magy. Kém. Foly. 125, 100 (2019) PDF
G. Czakó, T. Győri, B. Olasz, D. Papp, I. Szabó, V. Tajti, and D. A. Tasi
Benchmark ab initio and dynamical characterization of the stationary points of reactive atom + alkane and SN2 potential energy surfaces
Phys. Chem. Chem. Phys. 22, 4298 (2020) Perspective Cover PDF
T. Győri and G. Czakó
Automating the development of high-dimensional reactive potential energy surfaces with the ROBOSURFER program system
J. Chem. Theory Comput. 16, 51 (2020) PDF Highly Cited Paper (as of May/June 2020)
G. Avila, D. Papp, G. Czakó, and E. Mátyus
Exact quantum dynamics background of dispersion interactions: case study for CH4.Ar in full (12) dimensions
Phys. Chem. Chem. Phys. 22, 2792 (2020) PDF 2020 HOT PCCP article
D. A. Tasi, T. Győri, and G. Czakó
On the development of a gold-standard potential energy surface for the OH− + CH3I reaction
Phys. Chem. Chem. Phys. 22, 3775 (2020) Communication PDF 2020 HOT PCCP article
D. Papp, V. Tajti, T. Győri, and G. Czakó
Theory finally agrees with experiment for the dynamics of the Cl + C2H6 reaction
J. Phys. Chem. Lett. 11, 4762 (2020) PDF A Hónap Publikációja (MTA Kémiai Tudományok Osztálya)
E. M. Orján, A. B. Nacsa, and G. Czakó
Conformers of dehydrogenated glycine isomers
J. Comput. Chem. 41, 2001 (2020) PDF
B. Gruber and G. Czakó
Benchmark ab initio characterization of the abstraction and substitution pathways of the OH + CH4/C2H6 reactions
Phys. Chem. Chem. Phys. 22, 14560 (2020) PDF
P. Papp, V. Tajti, and G. Czakó
Numerical separation of the front-side attack and double-inversion retention pathways of SN2 reactions
Chem. Phys. Lett. 755, 137780 (2020) PDF
D. Papp and G. Czakó
Full-dimensional MRCI-F12 potential energy surface and dynamics of the F(2P3/2) + C2H6 __> HF + C2H5 reaction
J. Chem. Phys. 153, 064305 (2020) PDF
P. Papp and G. Czakó
Rotational mode specificity in the F− + CH3I(v=0, JK) SN2 and proton-transfer reactions
J. Phys. Chem. A 124, 8943 (2020) PDF
G. Czakó, T. Győri, D. Papp, V. Tajti, and D. A. Tasi
First-principles reaction dynamics beyond six-atom systems
J. Phys. Chem. A 125, 2385 (2021) Perspective Cover PDF
D. Papp and G. Czakó
Facilitated inversion complicates the stereodynamics of an SN2 reaction at nitrogen center
Chem. Sci. 12, 5410 (2021) Cover PDF
A. B. Nacsa and G. Czakó
Benchmark ab initio proton affinity of glycine
Phys. Chem. Chem. Phys. 23, 9663 (2021) Cover PDF
T. Szűcs and G. Czakó
Benchmark ab initio stationary-point characterization of the complex potential energy surface of the multi-channel Cl + CH3NH2 reaction
Phys. Chem. Chem. Phys. 23, 10347 (2021) PDF
D. A. Tasi, C. Tokaji, and G. Czakó
A benchmark ab initio study of the complex potential energy surfaces of the OH− + CH3CH2Y [Y = F, Cl, Br, I] reactions
Phys. Chem. Chem. Phys. 23, 13526 (2021) PDF 2021 HOT PCCP article
J. Meyer, V. Tajti, E. Carrascosa, T. Győri, M. Stei, T. Michaelsen, B. Bastian, G. Czakó, and R. Wester
Atomistic dynamics of elimination and nucleophilic substitution disentangled for the F− + CH3CH2Cl reaction
Nat. Chem. 13, 977 (2021) PDF A Hónap Publikációja (MTA Kémiai Tudományok Osztálya) Az Év Publikációja (Szegedi Tudományegyetem)
D. Papp, J. Li, H. Guo, and G. Czakó
Vibrational mode-specificity in the dynamics of the Cl + C2H6 __> HCl + C2H5 reaction
J. Chem. Phys. 155, 114303 (2021) PDF
V. Tajti, T. Győri, and G. Czakó
Detailed quasiclassical dynamics of the F− + CH3Br reaction on an ab initio analytical potential energy surface
J. Chem. Phys. 155, 124301 (2021) PDF
D. Papp and G. Czakó
Vibrational mode-specific dynamics of the F(2P3/2) + C2H6 __> HF + C2H5 reaction
J. Chem. Phys. 155, 154302 (2021) PDF
D. A. Tasi and G. Czakó
Uncovering an oxide ion substitution for the OH− + CH3F reaction
Chem. Sci. 12, 14369 (2021) Cover PDF 2021 Chemical Science HOT Article Collection
A. Á. Dékány, G. Z. Kovács, and G. Czakó
High-level systematic ab initio comparison of carbon- and silicon-centered SN2 reactions
J. Phys. Chem. A 125, 9645 (2021) PDF
A. Á. Dékány and G. Czakó
Benchmark ab initio proton affinity and gas-phase basicity of α-alanine based on coupled-cluster theory and statistical mechanics
J. Comput. Chem. 43, 19 (2022) PDF
Z. Kerekes, D. A. Tasi, and G. Czakó
SN2 reactions with an ambident nucleophile: A benchmark ab initio study of the CN− + CH3Y [Y = F, Cl, Br, and I] systems
J. Phys. Chem. A 126, 889 (2022) PDF
T. Győri and G. Czakó
ManyHF: A pragmatic automated method of finding lower-energy Hartree−Fock solutions for potential energy surface development
J. Chem. Phys. 156, 071101 (2022) Communication Cover PDF Featured
V. Tajti and G. Czakó
Vibrational mode-specific dynamics of the F− + CH3CH2Cl multi-channel reaction
Phys. Chem. Chem. Phys. 24, 8166 (2022) PDF
D. Papp and G. Czakó
Rotational mode-specificity in the Cl + C2H6 __> HCl + C2H5 reaction
J. Phys. Chem. A 126, 2551 (2022) PDF
P. Tóth, T. Szűcs, and G. Czakó
Benchmark ab initio characterization of the abstraction and substitution pathways of the Cl + CH3CN reaction
J. Phys. Chem. A 126, 2802 (2022) PDF
D. A. Tasi and G. Czakó
Unconventional SN2 retention pathways induced by complex formation: High-level dynamics investigation of the NH2− + CH3I polyatomic reaction
J. Chem. Phys. 156, 184306 (2022) PDF
B. Gruber, V. Tajti, and G. Czakó
Full-dimensional automated potential energy surface development and dynamics for the OH + C2H6 reaction
J. Chem. Phys. 157, 074307 (2022) PDF
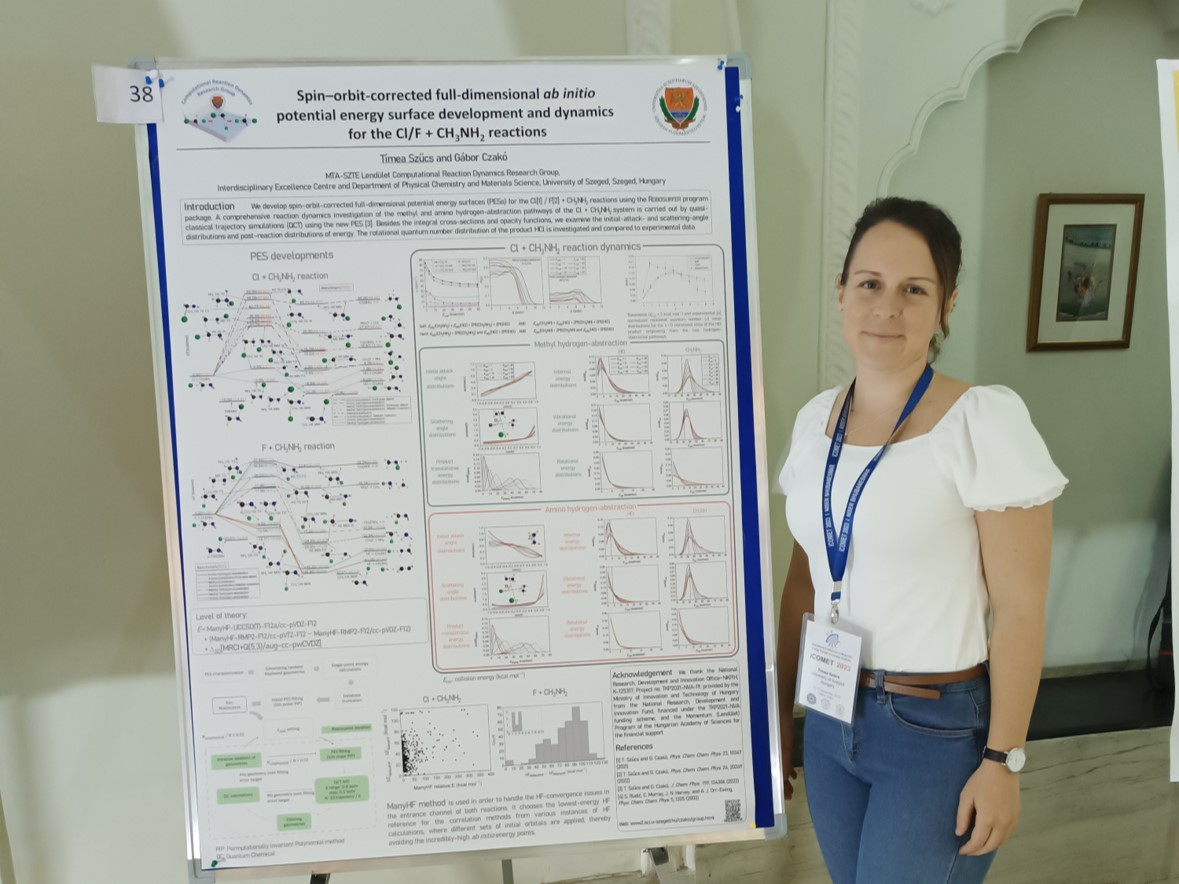
T. Szűcs and G. Czakó
Benchmark ab initio potential energy surface mapping of the F + CH3NH2 reaction
Phys. Chem. Chem. Phys. 24, 20249 (2022) PDF
C. Yin, V. Tajti, and G. Czakó
Full-dimensional potential energy surface development and dynamics for the HBr + C2H5 __> Br(2P3/2) + C2H6 reaction
Phys. Chem. Chem. Phys. 24, 24784 (2022) PDF 2022 HOT PCCP article
C. Yin and G. Czakó
Automated full-dimensional potential energy surface development and quasi-classical dynamics for the HI(X1Σ+) + C2H5 __> I(2P3/2) + C2H6 reaction
Phys. Chem. Chem. Phys. 24, 29084 (2022) PDF
A. B. Nacsa and G. Czakó
Benchmark ab initio determination of the conformers, proton affinities, and gas-phase basicities of cysteine
J. Phys. Chem. A 126, 9667 (2022) PDF
D. Papp, V. Tajti, G. Avila, E. Mátyus, and G. Czakó
CH4.F− revisited: full-dimensional ab initio potential energy surface and variational vibrational states
Mol. Phys. 121, e2113565 (2023) PDF
C. Yin and G. Czakó
Theoretical vibrational mode-specific dynamics studies for the HBr + C2H5 reaction
Phys. Chem. Chem. Phys. 25, 3083 (2023) PDF
D. A. Tasi, T. Michaelsen, R. Wester, and G. Czakó
Quasi-classical trajectory study of the OH− + CH3I reaction: Theory meets experiment
Phys. Chem. Chem. Phys. 25, 4005 (2023) PDF
T. Győri and G. Czakó
A comprehensive benchmark ab initio survey of the stationary points and products of the OH· + CH3OH system
J. Chem. Phys. 158, 034301 (2023) PDF
B. Gruber and G. Czakó
High-level ab initio mapping of the multiple H-abstraction pathways of the OH + glycine reaction
Phys. Chem. Chem. Phys. 25, 5271 (2023) PDF
A. B. Nacsa, M. Kígyósi, and G. Czakó
Protonation of serine: Conformers, proton affinities and gas-phase basicities at the "gold standard" and beyond
Phys. Chem. Chem. Phys. 25, 8891 (2023) PDF
C. Yin and G. Czakó
Vibrational mode-specific quasi-classical trajectory studies for the two-channel HI + C2H5 reaction
Phys. Chem. Chem. Phys. 25, 9944 (2023) PDF
A. B. Nacsa, V. Tajti, and G. Czakó
Dynamics of the Cl− + CH3I reaction on a high-level ab initio analytical potential energy surface
J. Chem. Phys. 158, 194306 (2023) PDF
A. Á. Dékány and G. Czakó
Exploring the versatile reactivity of the F− + SiH3Cl system on a full-dimensional coupled-cluster potential energy surface
J. Chem. Phys. 158, 224303 (2023) PDF
T. Gstir, T. Michaelsen, B. A. Long, A. B. Nacsa, A. Ayasli, D. Swaraj, F. Zappa, F. Trummer, S. G. Ard, N. S. Shuman, G. Czakó, A. A. Viggiano, and R. Wester
The influence of fluorination on the dynamics of the F− + CH3CH2I reaction
Phys. Chem. Chem. Phys. 25, 18711 (2023) PDF 2023 HOT PCCP article
C. Yin and G. Czakó
Competition between the H-abstraction and the X-abstraction pathways in the HX (X = Br, I) + C2H5 reactions
Phys. Chem. Chem. Phys. 25, 20241 (2023) PDF 2023 HOT PCCP article
A. Giricz, G. Czakó, and D. Papp
Alternating stereospecificity upon central-atom change: Dynamics of the F− + PH2Cl SN2 reaction compared to its C- and N-centered analogues
Chem. Eur. J. 29, e202302113 (2023) PDF A Hónap Publikációja (MTA Kémiai Tudományok Osztálya)
B. Gruber, V. Tajti, and G. Czakó
Vibrational mode-specific dynamics of the OH + C2H6 reaction
J. Phys. Chem. A 127, 7364 (2023) PDF
T. Szűcs and G. Czakó
ManyHF-based full-dimensional potential energy surface development and quasi-classical dynamics for the Cl + CH3NH2 reaction
J. Chem. Phys. 159, 134306 (2023) PDF
C. Yin and G. Czakó
Full-dimensional automated potential energy surface development and detailed dynamics for the CH2OO + NH3 reaction
Phys. Chem. Chem. Phys. 25, 26917 (2023) PDF
B. Ballay, T. Szűcs, D. Papp, and G. Czakó
Phosphorus-centered ion-molecule reactions: benchmark ab initio characterization of the potential energy surfaces of the X− + PH2Y [X, Y = F, Cl, Br, I] systems
Phys. Chem. Chem. Phys. 25, 28925 (2023) PDF
D. A. Tasi and G. Czakó
Vibrational mode-specificity in the dynamics of the OH− + CH3I multi-channel reaction
J. Chem. Phys. 160, 044305 (2024) PDF
T. Szűcs and G. Czakó
Automated potential energy surface development and comprehensive dynamics for the F + CH3NH2 reaction
J. Chem. Phys. 160, 064304 (2024) PDF Editor's Pick
A. B. Nacsa, C. Tokaji, and G. Czakó
High-level analytical potential-energy-surface-based dynamics of the OH− + CH3CH2Cl SN2 and E2 reactions in full (24) dimensions
Faraday Discuss. 251, 604 (2024) PDF
A. Á. Dékány and G. Czakó
Detailed quasiclassical dynamics of the F− + SiH3Cl multi-channel reaction
Phys. Chem. Chem. Phys. 26, 10008 (2024) PDF
A. Ayasli, P. Tóth, T. Michaelsen, T. Gstir, F. Zappa, D. Papp, G. Czakó, and R. Wester
Imaging the ion-molecule reaction dynamics of O− + CD4
J. Phys. Chem. A 128, 3078 (2024) PDF
G. Czakó, B. Gruber, D. Papp, V. Tajti, D. A. Tasi, and C. Yin
First-principles mode-specific reaction dynamics
Phys. Chem. Chem. Phys. 26, 15818 (2024) Perspective (PCCP 25th Anniversary Collection) Cover PDF 2024 HOT PCCP article
D. A. Tasi and G. Czakó
Benchmark ab initio characterization of the complex potential energy surfaces of the HOO− + CH3Y [Y = F, Cl, Br, I] reactions
Phys. Chem. Chem. Phys. 26, 16048 (2024) PDF
K. Horváth, V. Tajti, D. Papp, and G. Czakó
Dynamics of the HCl + C2H5 multi-channel reaction on a full-dimensional ab initio potential energy surface
J. Phys. Chem. A 128, 4474 (2024) PDF
D. R. Gál, D. Papp, and G. Czakó
Benchmark ab initio characterization of the multi-channel Cl + CH3X [X = F, Cl, Br, I] reactive potential energy surfaces
Phys. Chem. Chem. Phys. 26, 17695 (2024) PDF
C. Yin and G. Czakó
Revealing new pathways for the reaction of Criegee intermediate CH2OO with SO2
Commun. Chem. 7, 157 (2024) PDF
P. Tóth, T. Szűcs, T. Győri, and G. Czakó
Dynamics of the Cl + CH3CN reaction on an automatically-developed full-dimensional ab initio potential energy surface
J. Chem. Phys. 161, 084304 (2024) PDF
E. Mátyus and G. Czakó
Foreword to the Festschrift in honour of Professor Attila G. Császár: molecules (always) in motion
Mol. Phys. 122, e2360850 (2024) PDF
G. Alexandrowicz, D. Babikov, M. Brouard, A. Butler, H. Chadwick, D. W. Chandler, M. Fárník, J. Fingerhut, H. Guo, T. Győri, C. T. Haakansson, D. J. Harding, D. Heard, B. R. Heazlewood, D. Heathcote, N. Hertl, P. G. Jambrina, G.-J. Kroes, O. A. Krohn, P. D. Lane, V. Le Duc, H. J. Lewandowski, J. Loreau, M. McCrea, K. G. McKendrick, J. Meyer, D. R. Moon, A. S. Mullin, G. M. Nathanson, D. M. Neumark, K.-K. Ni, N. Pal, E. Pluhařová, C. Reilly, P. Robertson, S. J. Sibener, C. Sparling, V. Sridurai, A. Srivastav, M. Strutton, A. G. Suits, J. Wagner, P. D. Watson, R. Wester, S. Willitsch, A. M. Wodtke, and B. S. Zhao
Scattering in extreme environments: general discussion
Faraday Discuss. 251, 171 (2024) PDF
D. Babikov, N. Balucani, A. Bergeat, M. Brouard, D. W. Chandler, M. L. Costen, M. Fárník, H. Guo, T. Győri, D. Heard, D. Heathcote, N. Hertl, P. G. Jambrina, N. M. Kidwell, O. A. Krohn, V. Le Duc, J. Loreau, S. R. Mackenzie, M. McCrea, K. G. McKendrick, J. Meyer, D. R. Moon, A. S. Mullin, G. S. Nathanson, D. M. Neumark, K.-K. Ni, M. J. Paterson, E. Pluhařová, P. Robertson, C. Reilly, G. C. Schatz, C. Sparling, A. G. Suits, P. D. Watson, R. Wester, S. Willitsch, and A. M. Wodtke
Scattering of larger molecules – part 1: general discussion
Faraday Discuss. 251, 313 (2024) PDF
F. J. Aoiz, N. Balucani, A. Bergeat, A. Butler, D. W. Chandler, G. Czakó, T. Győri, D. E. Heard, D. Heathcote, B. R. Heazlewood, N. Hertl, P. G. Jambrina, R. I. Kaiser, O. A. Krohn, V. Le Duc, J. Loreau, S. R. Mackenzie, K. G. McKendrick, J. Meyer, G. M. Nathanson, D. M. Neumark, R. Pandey, C. Reilly, P. Robertson, G. C. Schatz, S. J. Sibener, A. G. Suits, P. D. Watson, R. Wester, S. Willitsch, A. M. Wodtke, and B. S. Zhao
Scattering of larger molecules – part 2: general discussion
Faraday Discuss. 251, 622 (2024) PDF
B. Gruber and G. Czakó
High-level ab initio characterization of the OH + CH3NH2 reaction
Phys. Chem. Chem. Phys. 26, 28543 (2024) PDF
B. J. Molnár, A. Á. Dékány, and G. Czakó
Automated potential energy surface development and quasi-classical dynamics for the F− + SiH3I system
J. Chem. Phys. 161, 194306 (2024) PDF
D. A. Tasi, E. M. Orján, and G. Czakó
Benchmark ab initio mapping of the F− + CH2ClI SN2 and proton-abstraction reactions
J. Phys. Chem. A 128, 10568 (2024) PDF
A. Ayasli, A. Khan, T. Gstir, T. Michaelsen, D. Papp, Y. Wang, H. Song, M. Yang, G. Czakó, and R. Wester
A dynamic isotope effect in the nucleophilic substitution reaction between F− and CD3I
Nat. Commun. 16, 2318 (2025) PDF
Y. Wang, Z. Tu, G. Czakó, H. Song, and M. Yang
Mode-specific quantum and quasi-classical trajectory dynamics of the F− + CH3I __> I− + CH3F SN2 reaction
J. Phys. Chem. A 129, 6344 (2025) PDF
A. Sunaga, T. Győri, G. Czakó, and E. Mátyus
Exact quantum dynamics of methanol: Full-dimensional ab initio potential energy surface of spectroscopic quality and variational vibrational states
J. Chem. Phys. 163, 064101 (2025) PDF
P. Tóth and G. Czakó
Vibrational mode-specific dynamics of the Cl + CH3CN reaction
J. Chem. Phys. 163, 064306 (2025) PDF
G. Czakó
First-principles dynamics and mechanisms of fundamental chemical reactions
Adv. Quantum Chem. megjelenés alatt (2026) Könyvfejezet
B. Gruber and G. Czakó
Site- and conformer-specific reaction dynamics of glycine with the hydroxyl radical
Commun. Chem. 9, 16 (2026) PDF
A. Á. Dékány, B. J. Molnár, and G. Czakó
Mode-specific quasi-classical dynamics of the F− + SiH3Cl system
Phys. Chem. Chem. Phys. DOI: 10.1039/D5CP04412J (2026)
Hírek
2026. február 7.
Attila és Balázs mód-specifikus F− + SiH3Cl cikkét elfogadja a PCCP.2026. január 12.
FM sikeresen megvédi BSc szakdolgozatát.2025. december 18.
Bohr Anna (BSc hallgató) csatlakozik a csoportunkhoz.2025. december 15.
Gábort egyetemi tanárrá nevezik ki.2025. december 6.
Gábor, Balázs és Petra előadást tart Mátrafüreden az MTA Anyag- és Molekulaszerkezeti Munkabizottság ülésén.2025. november 21.
Csaba előadást tart Mátrafüreden az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2025. november 20.
Petra és Tibi előadást tart Mátrafüreden az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2025. november 20.
Balázs cikkét az OH + glicin reakció funkciós-csoport- és konformer-specifikus dinamikájáról elfogadja a Communications Chemistry.2025. október 3.
Attila sikeresen megvédi PhD disszertációját (lásd Sajtó).2025. szeptember 8.
Sárik Levente (MSc hallgató) és Jancsó István (BSc hallgató) csatlakozik a csoporthoz.2025. szeptember 3.
Tibi contributed előadást tart Szegeden az International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) konferencián.2025. szeptember 1.
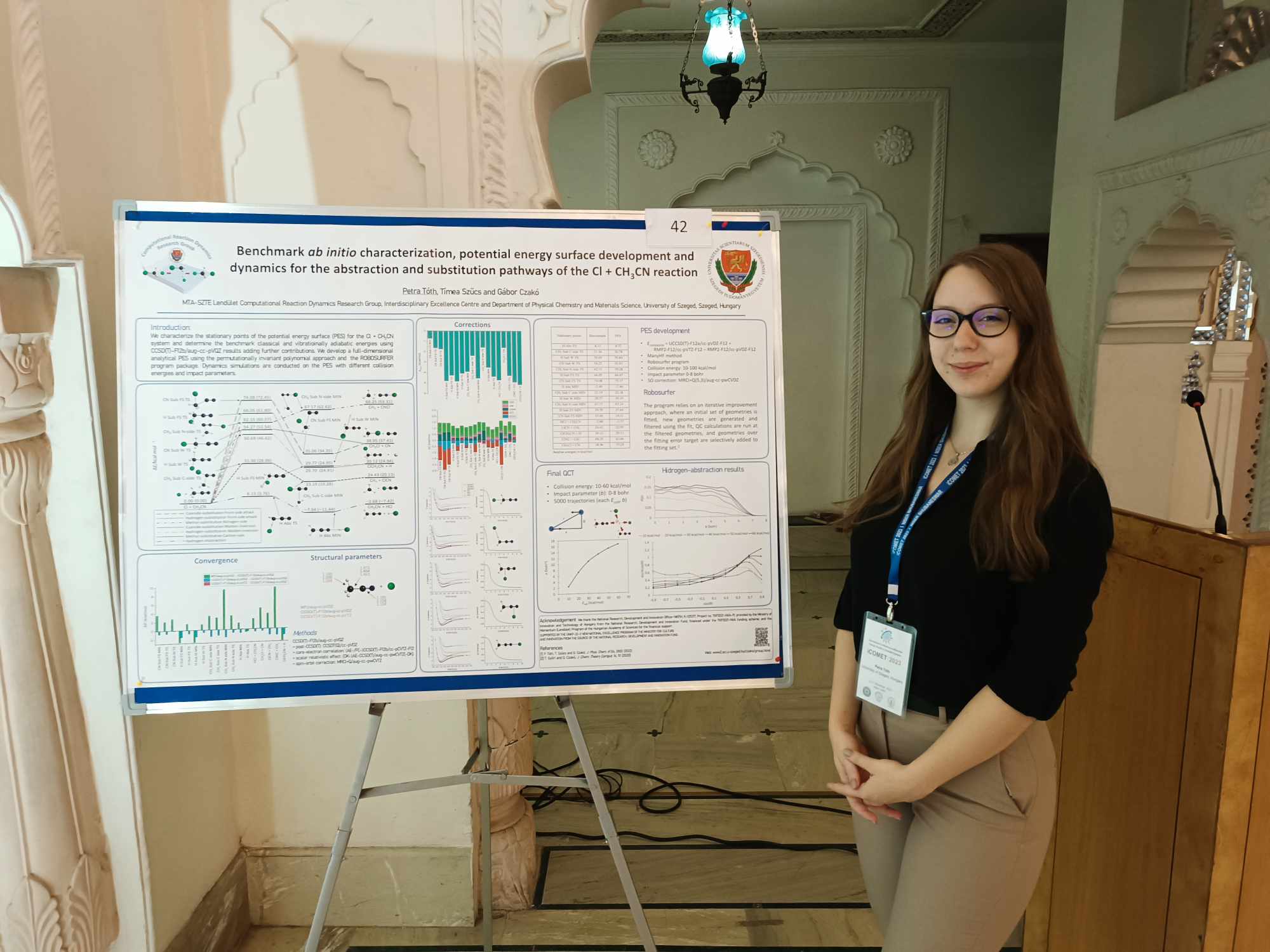
Gruber Balázs, Molnár Balázs, Petra és Viktor bemutatják a poszterüket az International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) konferencián (Szeged).2025. szeptember 1.
Petra hot topic előadást tart Szegeden az International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) konferencián.2025. szeptember 1.
Petra elnyeri az EKÖP ösztöndíjat.2025. augusztus 31.
Gábor megnyitja a Szegeden megrendezett International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) konferenciát.2025. augusztus 22.
Gábor előadásokat tart az ELTE PHYMOL Training School rendezvényén.2025. július 18.
Petra mód-specifikus Cl + CH3CN cikkét elfogadja a J. Chem. Phys.2025. július 16.
Tibi – Ayaki Sunagaval és Mátyus Edittel együttműködésben készült − cikkét a metanol PES-éről és rezgési állapotairól elfogadja a J. Chem. Phys.2025. július 7-8.
Petra bemutatja a poszterét a Dynamics of Molecular Collisions (DMC) konferencián (Snowbird, Utah, USA).2025. június 27.
Balázs, Tibi és Timi átveszik PhD okleveleiket.2025. június 26.
Gábor – Yan Wanggal és munkatársaival együttműködésben készült − cikkét a F− + CH3I SN2 reakció kvantum- és QCT-dinamikájáról elfogadja a J. Phys. Chem. A.2025. június 23.
Anett sikeresen megvédi BSc szakdolgozatát.2025. június 4.
Kitti sikeresen megvédi MSc diplomamunkáját.2025. május 29.
Tibi, Petra és Balázs előadást tartanak a KeMoMo-QSAR szimpóziumon.2025. április 24.
Megjelenik a hír a Nature Communications cikkünkről az SZTE honlapján (lásd Sajtó)!2025. április 11.
Dorina 3. díjat és egy különdíjat kap az OTDK-n.2025. április 10.
Dorina, Laura és Réka megtartják előadásaikat az OTDK-n.2025. április 9.
Balázs, Csaba és Dorina megtartják előadásaikat az OTDK-n.2025. március 28.
Balázs sikeresen megvédi PhD disszertációját.2025. február 13.
Megjelenik egy interjú a Lendület pályázat elnyerése kapcsán az MTA honlapján (lásd Sajtó)!2025. február 11.
Dóri − a Wester és Yang csoportokkal együttműködésben készült − cikkét egy dinamikai izotópeffektusról a F− + CD3I reakcióban elfogadja a Nature Communications!2025. február 7.
Timi sikeresen megvédi PhD disszertációját.2025. február 6.
Péter Panka (BSc hallgató) csatlakozik a csoportunkhoz.2025. február 5.
Szabadszállási Noémi (BSc hallgató) csatlakozik a csoportunkhoz.2025. január 29.
Kószó Lili (BSc hallgató) csatlakozik a csoportunkhoz.2025. január 16.
Tibi sikeresen megvédi PhD disszertációját.2025. január 16.
Gábor könyvfejezetét elfogadja az Advances in Quantum Chemistry.2025. január 8.
Balázs sikeresen megvédi BSc szakdolgozatát.2024. december 17.
Dóri elnyeri az NKFIH Starting kutatási pályázatát!2024. november 29.
Gábor átveszi a Lendület oklevelet és egy rövid előadást tart az MTA Lendület Nap rendezvényén.2024. november 28.
Dorina első és Balázs második díjat nyer a házi TDK konferencián.2024. november 22.
Gábor előadást tart Mátrafüreden az MTA Anyag- és Molekulaszerkezeti Munkabizottság ülésén.2024. november 15.
Domonkos és Erik F− + CH2ClI benchmark cikkét elfogadja a J. Phys. Chem. A.2024. november 7.
Petra és Dorina előadást tart Mátrafüreden az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2024. október 31.
Balázs és Attila cikkét a F− + SiH3I reakció PES-éről és dinamikájáról elfogadja a J. Chem. Phys.2024. október 31.
András sikeresen megvédi PhD disszertációját.2024. október 31.
Laura és Bálint előadást tart a Kémiai Előadói Napok konferencián.2024. október 30.
Kutatócsoportunk a Szeged TV Kvantum műsorában (lásd Sajtó)!2024. október 30.
Dorina és Réka előadást tart a Kémiai Előadói Napok konferencián.2024. október 29.
Petra díszelőadás tart a Kémiai Előadói Napokon és átveszi a Magyar Kémikusok Egyesülete Nívódíját.2024. október 28.
Balázs benchmark cikkét az OH + CH3NH2 reakcióról elfogadja a PCCP.2024. október 18.
Petra diplomamunkája elnyeri a Magyar Kémikusok Egyesülete Nívódíját.2024. szeptember 24.
Gábor meghívott előadást tart Pescarában a Molecular Electronic Structure (MES) konferencián.2024. szeptember 4.
Petra és Dorina elnyeri az EKÖP ösztöndíjat.2024. augusztus 23.
Mátyus Edit és Gábor szerkesztésével készült "Festschrift in honour of Prof. Attila G. Császár" megjelenik a Molecular Physics folyóiratban.2024. augusztus 12.
Petra, Timi és Tibi cikkét a Cl + CH3CN reakció PES-éről és dinamikájáról elfogadja a J. Chem. Phys.2024. július 15-17.
Roland Wester és négyen a csoportjából (Fabio, Christian, Dasarath és Jerin) meglátogatnak bennünket az Innsbruck-i Egyetemről.2024. július 13.
Az Origo.hu szintén leközli a "lendületes" hírt!2024. július 9.
Megjelenik a hír az új Lendület pályázatról az SZTE honlapján (lásd Sajtó)!2024. július 2.
Petra sikeres felvételi vizsgát tesz a Doktori Iskolába.2024. június 27.
Cangtao Criegee cikkét a CH2OO + SO2 új reakcióútjainak feltárásáról elfogadja a Communications Chemistry.2024. június 26.
Dóri, Petra és Viktor bemutatják a poszterüket Isztambulban a Quantum Reactive Scattering (QRS) konferencián.2024. június 25.
Gábor meghívott előadást tart Isztambulban a Quantum Reactive Scattering (QRS) konferencián.2024. június 18.
Petra átveszi az arany fokozatú SZTE Talent ösztöndíjat és az EPAM vállalati ösztöndíjat.2024. június 18.
Timi bemutatja PhD téziseit a Tanszéki Szemináriumon.2024. június 17.
Csaba sikeresen megvédi BSc szakdolgozatát.2024. június 14.
Laura, Dorina, Réka és Bálint sikeresen megvédik BSc szakdolgozatukat.2024. június 13.
Petra előadást tart az intézeti online ÚNKP konferencián.2024. június 12.
Megvan az újabb Lendület!2024. június 3.
Balázs bemutatja PhD téziseit, valamint Domonkos és Dóri előadást tartanak a legújabb eredményeikről a KeMoMo-QSAR szimpóziumon.2024. június 1.
Dorina és Dóri benchmark cikkét a Cl + CH3X [X = F, Cl, Br, I] reakciókról elfogadja a PCCP.2024. május 31.
Petra sikeresen megvédi MSc diplomamunkáját.2024. május 30.
Dóri és Timi előadást tartanak Balatonvilágoson az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2024. május 15-17.
Roland Thissen és David Lauvergnat meglátogatnak bennünket a Paris-Saclay Egyetemről.2024. május 15.
Kitti, Viktor és Dóri cikkét a HCl + C2H5 reakció PES-éről és dinamikájáról elfogadja a J. Phys. Chem. A.2024. május 10.
Tibi poszter díjat kap a New directions in molecular scattering Faraday Discussion konferencián (Edinburgh, Egyesült Királyság).2024. május 10.
Gábor egy online előadást tart a New directions in molecular scattering Faraday Discussion konferencián (Edinburgh, Egyesült Királyság).2024. május 8.
Balázs és Tibi bemutatják a poszterüket a New directions in molecular scattering Faraday Discussion konferencián (Edinburgh, Egyesült Királyság).2024. május 7.
Tarolunk az intézeti TDK konferencián: Csaba 1. díj, Dorina 1. díj, Laura 1. díj, Réka 2. díj és Bálint 3. díj!2024. május 6.
Domonkos benchmark cikkét a HOO− + CH3Y [Y = F, Cl, Br, I] reakciókról elfogadja a PCCP.2024. május 3.
András bemutatja PhD téziseit a Tanszéki−KeMoMo Szemináriumon.2024. április 16.
Viktor elnyeri az Akadémia utazási támogatását.2024. április 4.
A felkérésre írt Perspective cikkünket a first-principles módspecifikus reakciódinamikáról elfogadják a PCCP 25th Anniversary Collection kötetbe.2024. március 26.
Petra és Dóri − a Wester csoporttal együttműködésben készült − O− + CD4 cikkét elfogadja a J. Phys. Chem. A.2024. március 5.
Attila cikkét a F− + SiH3Cl reakció részletes dinamikájáról elfogadja a PCCP.2024. március 1.
Gábor egy meghívott előadást tart az Európai Kémiai Társaság Szerves Kémiai Osztálya által szervezett "Highlighting Organic Chemistry in Hungary" Webinaron.2024. február 20.
András és Csenge cikkét a OH− + CH3CH2Cl reakció PES-éről és dinamikájáról elfogadja a Faraday Discussions.2024. február 12.
Dóri meghívott előadást tart a Cambridge-i Egyetem Lennard-Jones Centre Discussion Group előadássorozatában.2024. február 12.
Két BSc hallgató (Felföldi Márton és Major Anett) csatlakozik a csoporthoz.2024. január 19.
Timi F + CH3NH2 JCP cikkét kiemelik, mint Editor's Pick.2024. január 18.
Timi cikkét a F + CH3NH2 reakció PES-éről és dinamikájáról elfogadja a J. Chem. Phys.2024. január 17.
András elnyeri a CEEPUS Freemover Mobilitási Ösztöndíjat két hónapra (Innsbruck, Wester csoport).2024. január 4.
Csenge sikeresen megvédi MSc diplomamunkáját.2023. december 25.
Domonkos mód-specifikus OH− + CH3I cikkét elfogadja a J. Chem. Phys.2023. december 14.
2025-ben mi szervezzük az International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) konferenciát Szegeden!2023. december 12.
Viktor sikeresen megvédi PhD disszertációját.2023. december 5.
Anett és Dóri Chem. Eur. J. cikkét az MTA Kémiai Osztálya a hónap publikációjának választja.2023. december 4.
Domonkos sikeresen megvédi PhD disszertációját.2023. november 25.
Balázs előadást tart Mátrafüreden az MTA Anyag- és Molekulaszerkezeti Munkabizottság ülésén.2023. november 24.
Domonkos, András és Timi előadást tartanak Mátrafüreden az MTA Anyag- és Molekulaszerkezeti Munkabizottság ülésén.2023. november 24.
Petra átveszi a Nagypál István Ösztöndíjat.2023. november 21.
Támogatást nyerünk a Nemzeti Kutatási, Fejlesztési és Innovációs Hivataltól a következő 4 évre.2023. november 15.
Gábor egy meghívott előadást tart Jaipurban (India) az International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) konferencián.2023. november 13-15.
Timi és Petra bemutatják a poszterüket az International Conference on Molecular Energy Transfer in Complex Systems (iCOMET) konferencián (Shiv Vilas Resorts, Jaipur, India).2023. november 1.
Benne vagyunk a Kari Naptárban (lásd Galéria)!2023. október 19.
Petra előadást tart a Kémiai Előadói Napokon.2023. szeptember 29.
Boldi, Timi és Dóri benchmark cikkét az X− + PH2Y [X, Y = F, Cl, Br, I] reakciókról elfogadja a PCCP.2023. szeptember 25.
Cangtao Criegee cikkét a CH2OO + NH3 reakció PES-éről és dinamikájáról elfogadja a PCCP.2023. szeptember 6.
Timi cikkét a Cl + CH3NH2 reakció PES-éről és dinamikájáról elfogadja a J. Chem. Phys.2023. szeptember 5.
Petra elnyeri az ÚNKP ösztöndíjat.2023. augusztus 8.
Balázs és Viktor mód-specifikus OH + C2H6 cikkét elfogadja a J. Phys. Chem. A.2023. augusztus 8.
Anett és Dóri cikkét a F− + PH2Cl reakció PES-éről és dinamikájáról elfogadja a Chem. Eur. J.2023. augusztus 2.
Cangtao HX (X = Br, I) + C2H5 cikkét, valamint András F− + CH3CH2I/CF3CH2I cikkét kiválasztják a 2023 HOT PCCP article gyűjteménybe.2023. július 13.
Cangtao cikkét a H/X-absztrakció versengéséről a HX (X = Br, I) + C2H5 reakciókban elfogadja a PCCP.2023. július 10-11.
András és Balázs bemutatják a poszterüket a Dynamics of Molecular Collisions (DMC) konferencián (Snowbird, Utah, USA).2023. július 1.
Petra átveszi a Kar Kiváló Hallgatója díjat.2023. június 29.
Petra előadást tart az intézeti online ÚNKP konferencián.2023. június 27.
Tibi bemutatja a poszterét Pozsonyban a 17th International Congress of Quantum Chemistry (ICQC) konferencián.2023. június 13.
Kitti sikeresen megvédi BSc szakdolgozatát.2023. június 9.
Attila előadást tart Balatonvilágoson az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2023. június 8.
Timi előadást tart Balatonvilágoson az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2023. június 8.
Petra megkapja az ezüst fokozatú SZTE Talent ösztöndíjat.2023. június 7.
András − a Wester és Viggiano csoportokkal együttműködésben készült − F− + CH3CH2I/CF3CH2I cikkét elfogadja a PCCP.2023. június 6.
Tibi és Viktor bemutatják PhD téziseiket a Tanszéki Szemináriumon.2023. május 24.
Zsolt sikeresen megvédi MSc diplomamunkáját.2023. május 18.
Attila cikkét a F− + SiH3Cl reakció PES-éről és dinamikájáról elfogadja a J. Chem. Phys.2023. május 18.
Petra, Barnabás és Cangtao előadást tartanak a KeMoMo-QSAR szimpóziumon.2023. május 17.
Domonkos bemutatja PhD téziseit a Tanszéki Szemináriumon.2023. április 30.
András és Viktor cikkét a Cl− + CH3I reakció PES-éről és dinamikájáról elfogadja a J. Chem. Phys.2023. április 15.
Petra és Máté 2., valamint Anett 3. helyezést ér el az OTDK-n.2023. április 14.
Barnabás, Anett és Máté megtartják előadásaikat az OTDK-n.2023. április 13.
Zsolt, Petra és Csenge megtartják előadásaikat az OTDK-n.2023. március 7.
Cangtao mód-specifikus HI + C2H5 cikkét elfogadja a PCCP.2023. március 6.
András és Máté cikkét a szerin konformereiről, protonaffinitásáról és gázfázisú bázicitásáról elfogadja a PCCP.2023. február 1.
Takó Dominika (középiskolás kutatódiák) csatlakozik a csoporthoz.2023. január 17.
Balázs OH + glicin cikkét elfogadja a PCCP.2023. január 3.
Rudner Csaba (BSc hallgató) is csatlakozik hozzánk.2023. január 2.
Négy BSc hallgató (Erdei Laura, Gál Dorina, Gyimesi Réka és Imre Bálint) és három középiskolás kutatódiák (Baranyi Bartal, Fuder Péter és Kocsis Nándor) csatlakozik a csoporthoz.2022. december 20.
Tibi OH + CH3OH cikkét elfogadja a J. Chem. Phys.2022. december 16.
Gábor előadást tart Szentesen a Koszta József Általános Iskolában.2022. december 16.
Domonkos − a Wester csoporttal együttműködésben készült − cikkét az OH− + CH3I reakció dinamikájáról elfogadja a PCCP.2022. december 16.
Boldi sikeresen megvédi BSc szakdolgozatát.2022. december 15.
Cangtao mód-specifikus HBr + C2H5 cikkét elfogadja a PCCP.2022. december 1.
András cikkét a cisztein konformereiről, protonaffinitásáról és gázfázisú bázicitásáról elfogadja a J. Phys. Chem. A.2022. november 16.
Gábor előadást tart az MTA-SZAB Székházban a SZAB Kémiai Szakbizottságának előadónapján a Magyar Tudomány Ünnepe 2022. rendezvénysorozat keretében.2022. november 14.
Megjelenik egy interjú az Év Publikációja Díj kapcsán az SZTE honlapján (lásd Sajtó)!2022. november 8.
Cangtao cikkét a HI + C2H5 reakció PES-éről és dinamikájáról elfogadja a PCCP.2022. november 1.
Cangtao és Viktor HBr + C2H5 cikkét "2022 HOT PCCP article"-nek választják.2022. november 1.
Dóri meghívott előadást tart Krétán a Stereodynamics konferencián.2022. október 28.
Balázs előadást tart Mátrafüreden az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2022. október 27.
Dóri és András előadást tartanak Mátrafüreden az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2022. október 22.
András és Tibi előadást tartanak Mátrafüreden az MTA Anyag- és Molekulaszerkezeti Munkabizottság ülésén.2022. október 21.
Gábor előadást tart Mátrafüreden az MTA Anyag- és Molekulaszerkezeti Munkabizottság ülésén.2022. szeptember 20.
Cangtao és Viktor cikkét a HBr + C2H5 reakció PES-éről és dinamikájáról elfogadja a PCCP.2022. szeptember 8.
Domonkos előadást tart Balatonföldváron a Quantum Reactive Scattering (QRS) konferencián.2022. szeptember 5.
Tibi előadást tart Balatonföldváron a Quantum Reactive Scattering (QRS) konferencián.2022. szeptember 2.
Petra elnyeri az ÚNKP ösztöndíjat.2022. szeptember 1.
Gábor megkapja az Év Publikációja Díjat (Nature Chemistry cikk) a Szegedi Tudományegyetemen.2022. augusztus 8.
Dóri és Viktor − a Mátyus csoporttal (ELTE) együttműködésben készült − cikkét a CH4.F− PES-éről és variációs rezgési állapotairól elfogadja a Mol. Phys. Ez a ROBOSURFER első spektroszkópiai alkalmazása!2022. augusztus 8.
Gábor előadást tart a University of New Mexico tanszéki szemináriumán.2022. augusztus 2.
Timi F + CH3NH2 cikkét elfogadja a PCCP.2022. július 22.
Balázs és Viktor cikkét az OH + C2H6 reakció PES-éről és dinamikájáról elfogadja a J. Chem. Phys. Ez az első 10-atomos PES a csoportban!2022. július 13-14.
András, Dóri, Timi és Viktor bemutatják a poszterüket a Molecular Interactions and Dynamics GRC konferencián (Stonehill College, Easton, USA).2022. július 12.
Tibi előadást tart a Molecular Interactions and Dynamics GRC konferencián (Stonehill College, Easton, USA).2022. július 11-12.
Tibi bemutatja a poszterét a Molecular Interactions and Dynamics GRC konferencián (Stonehill College, Easton, USA).2022. július 11.
Gábor vezeti a Molecular Interactions and Collisions in the Gas Phase szekciót a Molecular Interactions and Dynamics GRC konferencián (Stonehill College, Easton, USA).2022. június 21.
Tibi egy meghívott előadást tart Osloban (Norvégia) a 3. "Global SCF Optimization" YoungCAS Workshop-on.2022. június 15.
Petra sikeresen megvédi BSc szakdolgozatát.2022. június 14.
Anett, Barnabás és Máté sikeresen megvédik BSc szakdolgozatukat.2022. június 9.
András, Balázs és Timi sikeres komplex doktori vizsgát tesznek.2022. június 2.
Attila, Domonkos és Tibi előadást tartanak a KeMoMo-QSAR szimpóziumon.2022. május 20.
Petra online előadást tart az MTA Reakciókinetikai és Fotokémiai Munkabizottság balatonvilágosi ülésén.2022. május 19.
Petra átveszi a bronz fokozatú SZTE Talent ösztöndíjat.2022. május 19.
Gábor és Anett online előadást tartanak az MTA Reakciókinetikai és Fotokémiai Munkabizottság balatonvilágosi ülésén.2022. május 10.
Domonkos elnyeri a Nemzet Fiatal Tehetségeiért Ösztöndíjat.2022. április 27.
Anett első, Barnabás, Máté, Petra és Zsolt második és Csenge harmadik díjat nyer a házi TDK konferencián.2022. április 15.
Domonkos cikkét az NH2− + CH3I reakció PES-éről és dinamikájáról elfogadja a J. Chem. Phys.2022. április 11.
Petra és Timi benchmark ab initio Cl + CH3CN cikkét elfogadja a J. Phys. Chem. A.2022. április 4.
Dóri forgási mód-specifikus Cl + C2H6(JK) cikkét elfogadja a J. Phys. Chem. A.2022. március 24.
Elindul az Ígéretes fiatal kémikusaink rovat a Magyar Kémikusok Lapjában, amely a mi csoportunk bemutatásával kezdődik. Sőt, a címlapon is mi vagyunk (lásd Sajtó)!2022. március 21.
Viktor mód-specifikus F− + CH3CH2Cl cikkét elfogadja a PCCP.2022. március 7.
Cangtao Yin (posztdoktor) és Ballay Boldizsár (BSc hallgató) csatlakozik a csoporthoz.2022. március 2.
Molnár Balázs (BSc hallgató) csatlakozik a csoportunkhoz.2022. február 15.
Tibi ManyHF cikke a JCP címlapján és a Featured cikkek között!2022. február 9.
Demes Sándor előadást tart a csoportszemináriumunkon.2022. január 31.
Demes Sándor vendégünk megérkezik a Université de Rennes 1 egyetemről.2022. január 20.
Tibi ManyHF cikkét Communication-ként elfogadja a J. Chem. Phys.2022. január 19.
Zsolt és Domonkos ambidens CN− + CH3Y cikkét elfogadja a J. Phys. Chem. A.2022. január 12.
Attila sikeres komplex doktori vizsgát tesz.2021. november 29.
A Nature Chemistry cikkünket az MTA Kémiai Osztálya a hónap publikációjának választja.2021. november 27.
Petra előadást tart a Móra Interdiszciplináris Konferencián.2021. november 22.
Petra előadást tart a Móra Szabadegyetemen.2021. november 13.
Gábor átveszi az Év Fiatal Kutatója elismerő oklevelet.2021. november 10.
Domonkos oxid ion szubsztitúciós cikke a Chemical Science borítóján és a "2021 Chemical Science HOT Article Collection"-ben!2021. november 10.
Domonkos elnyeri a Nemzet Fiatal Tehetségeiért Ösztöndíjat.2021. október 22.
Balázs, Domonkos és Timi előadást tartanak Mátrafüreden az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2021. október 21.
Dóri előadást tart Mátrafüreden az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2021. október 13.
Attila és Gyula cikkét a C- vs. Si-centrumú SN2 reakciókról elfogadja a J. Phys. Chem. A.2021. október 13.
Domonkos cikkét egy új oxid ion szubsztitúciós reakcióút felfedezéséről az OH− + CH3F reakció esetén elfogadja a Chem. Sci.2021. október 8.
Csenge elnyeri a Bay Zoltán tanulmányi ösztöndíjat Gyula Város Önkormányzatától.2021. október 6.
Gábor előadást tart a Pécsi Tudományegyetemen.2021. szeptember 30.
Dóri, Viktor és András előadást tartanak a KeMoMo-QSAR szimpóziumon.2021. szeptember 28.
Gábor egy online meghívott előadást tart a "Machine Learning and Informatics for Chemistry and Materials" Telluride Workshop-on.2021. szeptember 27.
Attila cikkét az alanin protonaffinitásáról elfogadja a J. Comput. Chem.2021. szeptember 24.
Dóri cikkét a F + C2H6 reakció mód-specifikus dinamikájáról elfogadja a J. Chem. Phys.2021. szeptember 1.
Horváth Kitti (BSc hallgató) csatlakozik csoportunkhoz.2021. augusztus 30.
Viktor és Tibi cikkét a F− + CH3Br reakció dinamikájáról elfogadja a J. Chem. Phys.2021. augusztus 28.
Dóri cikkét a Cl + C2H6 reakció mód-specifikus dinamikájáról elfogadja a J. Chem. Phys.2021. augusztus 12.
Az MTA honlapja is beszámol a Nature Chemistry cikkünkről (lásd Sajtó)!2021. augusztus 10.
Megjelenik a hír a Nature Chemistry cikkünkről az SZTE honlapján és a delmagyar.hu oldalon (lásd Sajtó)!2021. augusztus 9.
A Nature Chemistry cikkünk megjelenik online!2021. augusztus 2.
Tanszékünk Lendület csoportjai a delmagyar.hu oldalon (lásd Sajtó).2021. július 27.
Bemutatják a tanszékünk Lendület csoportjait az SZTE honlapján (lásd Sajtó).2021. július 6.
Domonkos és Csenge OH− + CH3CH2Y cikkét "2021 HOT PCCP article"-nek választják.2021. június 16.
Csenge sikeresen megvédi BSc szakdolgozatát.2021. június 15.
Viktor és Tibi cikkét elfogadja a Nature Chemistry!2021. június 15.
Zsolt sikeresen megvédi BSc szakdolgozatát.2021. május 31.
Demeter sikeresen megvédi MSc diplomamunkáját.2021. május 27.
Domonkos, Tibi és Attila előadást tartanak az MTA Reakciókinetikai és Fotokémiai Munkabizottság online ülésén.2021. május 24.
Domonkos és Csenge OH− + CH3CH2Y cikkét elfogadja a PCCP.2021. május 18.
Balázs előadást tart az OTDK konferencián.2021. április 28.
András glicines cikke a PCCP címlapján!2021. április 21.
Dóri multi-inverziós cikke a Chemical Science címlapján!2021. április 13.
Gábor előadást tart a 2021-es tavaszi ACS Meeting Bill Hase Memory szimpóziumán.2021. április 2.
Timi Cl + CH3NH2 cikkét elfogadja a PCCP.2021. április 1.
A Perspective cikkünk a JPCA címlapján és a legolvasottabb (Most Read) cikkek között!2021. március 29.
András cikkét a glicin protonaffinitásáról elfogadja a PCCP.2021. március 12.
Dóri cikkét a F− + NH2Cl reakció multi-inverziós mechanizmusának felfedezéséről elfogadja a Chem. Sci.2021. február 22.
Gábor portréja megjelenik az MTA honlapján a legsikeresebb Bolyai-ösztöndíjasokat bemutató sorozatban (lásd Sajtó).2021. február 16.
A Perspective cikkünket elfogadja a J. Phys. Chem. A.2021. február 3.
Kígyósi Máté (BSc hallgató) csatlakozik a csoportunkhoz.2021. január 4.
Gábor egy online meghívott előadást tart a Beijing Institute of Technology egyetemen.2020. november 11.
Viktor sikeres komplex doktori vizsgát tesz.2020. november 6.
Viktor előadást tart az MTA Reakciókinetikai és Fotokémiai Munkabizottság online ülésén.2020. október 2.
Paszkál cikkét a F− + CH3I(JK) reakció JK-függő dinamikájáról elfogadja a J. Phys. Chem. A.2020. szeptember 23.
Balázs első díjat nyer a házi TDK konferencián.2020. szeptember 15.
Tibi ROBOSURFER cikke "Highly Cited Paper" (top 1% a területen) lesz a Web of Science oldalon.2020. szeptember 1.
Balázs, András és Timi megkezdik PhD munkájukat, valamint három BSc hallgató (Giricz Anett, Sipos Barnabás és Tóth Petra) csatlakozik a csoporthoz.2020. augusztus 28.
Timi sikeres felvételi vizsgát tesz a Doktori Iskolába.2020. július 19.
Dóri cikkét a F + C2H6 reakció PES-éről és dinamikájáról elfogadja a J. Chem. Phys.2020. július 7.
Paszkál és Viktor cikkét az elölről támadásos és dupla inverziós SN2 reakcióutak szétválasztásáról elfogadja a Chem. Phys. Lett.2020. június 26.
András és Balázs sikeres felvételi vizsgát tesz a Doktori Iskolába.2020. június 22.
Balázs OH + CH4/C2H6 cikkét elfogadja a PCCP.2020. június 10.
Erik és András cikkét a dehidrogénezett glicin izomerek konformereiről elfogadja a J. Comput. Chem.2020. június 1.
Szűcs Tímea csatlakozik a csoportunkhoz.2020. május 28.
Balázs és Erik sikeresen megvédi MSc diplomamunkáját.2020. május 27.
Tibi sikeres komplex doktori vizsgát tesz.2020. május 22.
Dóri, Viktor és Tibi cikkét a Cl + C2H6 reakció PES-éről és dinamikájáról elfogadja a J. Phys. Chem. Lett.2020. május 22.
Anna sikeresen megvédi BSc szakdolgozatát.2020. április 6.
Két cikkünket (OH− + CH3I és CH4.Ar PES-ek) "2020 HOT PCCP article"-nek választják.2020. február 26.
Az összefoglaló "Perspective" cikkünk a PCCP címlapján!2020. február 21.
Elkészültek a felújított irodáink!2020. február 3.
Tibi ROBOSURFER cikke a legolvasottabb közlemény a JCTC folyóiratban az elmúlt 30 napban. Január 16-án az 5., majd január 27-én a 4. volt a legolvasottabb cikkek között.2020. február 3.
Dékány Attila (PhD hallgató), Papp Paszkál (MSc projektmunkás) és Traj Adrián (MSc projektmunkás) csatlakozik a csoporthoz.2020. január 27.
Domonkos és Tibi cikkét a OH− + CH3I reakció PES-éről Communication-ként elfogadja a PCCP.2020. január 14.
Szepesi Dorina (középiskolás kutatódiák) csatlakozik a csoportunkhoz.2019. december 20.
Dóri CH4.Ar cikkét, ami a Mátyus csoporttal (ELTE) együttműködésben készült, elfogadja a PCCP.2019. december 18.
Tibi ROBOSURFER cikkét elfogadja a JCTC.2019. december 6.
Balázs sikeresen megvédi PhD disszertációját.2019. december 6.
A Perspective cikkünket elfogadja a PCCP.2019. november 8.
Gábor előadást tart Mátrafüreden az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2019. november 7.
Domonkos előadást tart Mátrafüreden az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2019. november 6.
Dóri előadást tart az MTA-SZAB Székházban a "Fiatal kémikusok a tudomány szolgálatában" előadónapon.2019. október 30.
Domonkos előadást tart a Kémiai Előadói Napokon.2019. szeptember 19.
Gábor előadást tart az Elméleti Fizikai Tanszék szemináriumán.2019. szeptember 18.
Gábor az Eötvös Kémia Műhely meghívott előadója.2019. szeptember 5.
Kutatócsoportunk bemutatkozik a Szeged TV Kvantum műsorában.2019. július 18.
Tanszéki Lendület ünnepséget tartunk.2019. július 4.
Gábor meghívott előadást tart Tokióban a Quantum Reactive Scattering (QRS) konferencián.2019. július 3.
Dóri bemutatja poszterét Tokióban a Quantum Reactive Scattering (QRS) konferencián.2019. július 1.
Gábort egyetemi docensnek nevezik ki.2019. június 27.
Gábor elnyeri a Lendület program támogatását.2019. június 22.
Csoportunk öt tagja (Gábor, Dóri, Domonkos, Tibi és Viktor) megérkezik Edinburghba, ahol részt vesznek az International Symposium on Molecular Beams (ISMB) konferencián.2019. június 6.
Gábor előadást tart a KeMoMo-QSAR szimpóziumon.2019. május 30.
Tokaji Csenge (BSc hallgató) csatlakozik a csoportunkhoz.2019. május 30.
Domonkos sikeres komplex doktori vizsgát tesz.2019. május 27.
Demeter sikeresen megvédi BSc dolgozatát.2019. május 15.
Kerekes Zsolt (BSc hallgató) csatlakozik a csoportunkhoz.2019. május 14.
Balázs sikeresen megtartja PhD elővédését.2019. május 9.
Domonkos elismerő oklevelet vesz át a Talent ösztöndíj PhD kategóriájában.2019. május 6.
Balázs sikeres doktori szigorlatot tesz.2019. április 4.
Domonkos előadást tart Debrecenben az I. FKF szimpóziumon.2019. március 15.
Domonkos és Zita második SN2 reakciókról szóló cikkét elfogadja a PCCP.2019. február 4.
Schmidt Anna (BSc hallgató) csatlakozik a csoportunkhoz.2019. február 1.
Viktor megkezdi PhD munkáját.2018. december 20.
20:10-kor elfogadják Balázs JPCA cikkét. 22:33-kor elfogadják Balázs PCCP cikkét.2018. december 20.
Domonkos elnyeri a Nemzet Fiatal Tehetségeiért Ösztöndíjat.2018. december 14.
Viktor sikeresen megvédi MSc diplomamunkáját.2018. december 4.
Gábor és a Wester csoport közös cikkét az ArH+ és CO proton transzfer dinamikájáról elfogadja az Int. J. Mass Spectrom.2018. november 30.
Dóri és Gruber Balázs X + C2H6 [X = F, Cl, Br, I] reakciókról szóló cikkét elfogadja a Phys. Chem. Chem. Phys.2018. november 22.
Viktor második díjat nyer a házi TDK konferencián.2018. november 8.
Dóri, Domonkos, Viktor, Tibi és Balázs előadást tartanak Veszprémben az MTA Reakciókinetikai és Fotokémiai Munkabizottság ülésén.2018. október 16.
Balázs és Domonkos előadást tart a Kémiai Előadói Napok konferencián.2018. október 15.
Tibi diplomamunkája elnyeri a Magyar Kémikusok Egyesülete Nívódíját, amit a Kémiai Előadói Napokon egy díszelőadás után vesz át.2018. október 1.
Balázs elnyeri a Richter 6 hónapos ösztöndíját.2018. szeptember 19.
Balázs cikkét a F− + CH3I reakció mód-specifikus dinamikájáról elfogadja a J. Phys. Chem. A.2018. július 7.
Gábor átveszi a Természettudományi és Informatikai Kar Tudományos Díját.2018. június 29.
Gábor átveszi a habilitációs oklevelét a Szegedi Tudományegyetemen.2018. június 28.
Gábor Bolyai Plakett kitüntetést vesz át a Magyar Tudományos Akadémia elnökétől.2018. június 7.
Domonkos és Zita OH− + CH3Y [Y = F, Cl, Br, I] SN2 reakciókról szóló cikkét elfogadja a J. Phys. Chem. A.2018. június 4.
Tibi sikeresen megvédi MSc diplomamunkáját.2018. május 29.
András, Erik és Balázs sikeresen megvédi BSc dolgozatát.2018. május 24.
Balázs, Domonkos, Tibi és Viktor előadást tart a KeMoMo-QSAR 2018 szimpóziumon.2018. május 9.
Balázs nemzetközi együttműködésben készült cikkét elfogadja a Science Advances.2018. május 9.
Gábor előadást tart az MTA Székházban "A kvantumkémia Magyarországon" című ünnepi tudományos ülésen.2018. április 18.
Gábor Budapesten tart meghívott előadást az Anharmonicity in medium-sized molecules and clusters (AMOC) Meeting-en.2018. március 16.
Tibi első cikke megjelenik a J. Phys. Chem. A folyóiratban, amely úttörő-jellegű közlemény részletesen vizsgálja az elektronszerkezet-számítás szintjének hatását egy reakció dinamikájára.2018. február 27.
Gábor sikeres habilitációs előadást tart.2018. január 23.
Bálint N-centrumú SN2 reakciókat tárgyaló cikke megjelenik a J. Phys. Chem. A folyóiratban.2018. január 1.
Papp Dóra posztdoktori kutatóként csatlakozik a csoporthoz.2017. december 12.
Gábor egy meghívott előadást tart egy COST Workshop-on Ciudad Realban.2017. december 6.
Gábor átveszi az MTA doktori oklevelét és Ő lesz a Magyar Tudományos Akadémia legfiatalabb nagydoktora.2017. november 30.
Az szerkesztők felkérésére írt SN2 reakciókat tárgyaló összefoglaló cikkünket (Feature Article) a J. Phys. Chem. A folyóirat a címlapján emeli ki.2017. november 21.
Laci cikke, amely új reakcióutakat tár fel a Cl + CH4 -> H + CH3Cl reakcióra megjelenik a J. Phys. Chem. A folyóiratban.2017. október 29.
Gábor egy meghívott előadást tart egy, az ACS Southwest Regional Meeting-en megrendezett SN2 Reaction Dynamics Symposium-on Lubbock-ban, Texas államban.2017. szeptember 12.
Kopin Liu kísérleti csoportjával való együttműködés meghozta az eredményét! Gábornak újabb Nature Chemistry cikke jelenik meg a Cl + CHD3 reakció szögfüggő energiagátjáról.2017. szeptember 1.
Tasi Domonkos Attila (PhD hallgató), Gruber Balázs (BSc hallgató) és Orján Erik (BSc hallgató) csatlakozik csoportunkhoz.2017. július 13.
Gábor egy 4-éves NKFI pályázatot nyer.2017. július 10.
István Cl− + CH3I cikke megjelenik a J. Phys. Chem. A folyóiratban.2017. július 3.
Gábor egy meghívott előadást tart Triesztben a XIV International Workshop on Quantum Reactive Scattering konferencián.2017. június 29.
Zita sikeres előadást tart a projektmunka beszámolón.2017. június 20.
Gábor egy meghívott előadást tart Torunban (Lengyelország) a 8th International Meeting on Atomic and Molecular Physics and Chemistry (IMAMPC) konferencián.2017. június 9.
István és Balázs TOP cikke megjelenik a J. Phys. Chem. Lett. folyóiratban. A közlemény, egy új módszer, trajektória ortogonális projekció (TOP) segítségével rávilágít az elölről-támadásos komplex képződésének szerepére SN2 reakciókban.2017. május 26.
Gyula megvédi BSc értekezését.2017. március 24.
Viktor F− + CH3CH2Cl benchmark cikke megjelenik a J. Phys. Chem. A folyóiratban.2017. március 7.
Ván Demeter (BSc hallgató) csatlakozik a csoportunkhoz.2017. február 15.
A Chemical Science elfogadja Balázs első cikkét a F− + CH3I reakció potenciális energia felületéről és dinamikájáról.2017. február 1.
Krotos László (MSc hallgató) csatlakozik a csoportunkhoz.2017. január 26.
Bálint sikeres előadást tart a projektmunkájáról.2017. január 25.
Gábor megvédi az MTA doktori disszertációját.2016. december 22.
Viktor megvédi BSc értekezését.2016. november 24.
Tibi első díjat nyer a házi TDK konferencián.2016. október 3.
István cikke a F− + CHD2Cl reakció multi-csatornás, mód-specifikus dinamikájáról megjelenik a J. Chem. Phys. folyóiratban.2016. szeptember 1.
Sok új hallgató, Fábián Zita (MSc), Hajdu Bálint (MSc), Kovács Gyula (BSc), Nacsa András (BSc) és Tajti Viktor (BSc) csatlakozik a csoportunkhoz.2016. augusztus 9.
Megjelenik Minghui Yang és Hua Guo professzorokkal közös cikkünk a rangos J. Phys. Chem. Lett. folyóiratban, amely együttműködés az eddigi legpontosabb kvantumdinamika szimulációt eredményezte egy SN2 reakcióra.2016. július 12.
István megvédi a "Dynamics of biomolecular nucleophilic substitution and proton-transfer reactions on global ab initio potential energy surfaces" című PhD disszertációját. Munkáját a neves King's College London egyetemen folytatja posztdoktori ösztöndíjasként Rosta professzor csoportjában.2016. május 24.
Tibi megvédi a BSc értekezését és Gergő sikeres projektmunka beszámolót tart.2016. február 1.
Szekeres Gergő csatlakozik hozzánk, mint MSc projektmunkás.2015. november 30.
Megjelenik az innsbrucki Wester csoporttal együttműködésben készült Nature Chemistry cikkünk az SN2 reakciók meglepő távozócsoport effektusáról.2015. november 17.
Az új dupla-inverzós mechanizmust bemutató [Nature Communications 6, 5972 (2015)] cikkünk, amit korábban kiemelt a National Geographic Magyarország, az Index.hu és az MTVA "Highly Cited Paper" lesz a Web of Science oldalon.2015. szeptember 7.
Gábor egy meghívott előadást tart Besztercebányán a 14th Central European Symposium on Theoretical Chemistry (CESTC) konferencián.2015. szeptember 1.
Szabó István (tudományos segédmunkatárs), Olasz Balázs (PhD hallgató) és Győri Tibor (BSc hallgató) csatlakozik a csoporthoz.2015. augusztus 1.
Czakó Gábor egyetemi adjunktusként érkezik az SZTE-re, majd vezetésével megalakul az Elméleti Reakciódinamika Kutatócsoport.Sajtómegjelenések
A tudomány: művészet! – Először adták át az SZTE Kémia Doktori Iskola emlékérmet
SZTE honlap, az első éremátadás alkalmából Dékány Attila PhD védésén (2025)
Nature Communications cikkben írta le egy kémiai reakció kvantumos alagúthatását Dr. Papp Dóra és Dr. Czakó Gábor
SZTE honlap, a Nature Communications cikkünk megjelenése alkalmából (2025)
Lendületesek: Czakó Gábor
MTA honlap, az újabb Lendület pályázat elnyeréséről (2025)
Kutatócsoportunk a Szeged TV műsorában
Szeged TV Kvantum (7:43-tól), az újabb Lendület pályázat elnyerése kapcsán (2024)
Kvantumhatások az atomi terepasztalon - Dr. Czakó Gábor második MTA Lendület-pályázatát nyerte el
SZTE honlap, az újabb Lendület pályázat elnyeréséről (2024)
Dr. Czakó Gábor (SZTE) egyetemi docens: "A számításos kémia pontosabb lehet, mint a kísérlet"
SZTE honlap, az Év Publikációja Díj kapcsán (2022)
Elméleti reakciódinamika-kutatások a Szegedi Tudományegyetemen
Magyar Kémikusok Lapja, kutatócsoportunk bemutatása az Ígéretes fiatal kémikusaink rovatban (2022) Címlap
Lendületes kutatók elsőként azonosították egy kémiai reakció kísérleti ujjlenyomatát
MTA honlap, a 3. Nature Chemistry cikk megjelenése alkalmából (2021)
Összetett rendszer kémiai reakciójáról közöltek cikket szegedi kutatók
Delmagyar.hu, a 3. Nature Chemistry cikk megjelenése alkalmából (2021)
Vegyszerek nélküli kémia – új Nature Chemistry cikk az SZTE Czakó-csoportjától
SZTE honlap, a 3. Nature Chemistry cikk megjelenése alkalmából (2021)
Lendület: hat szegedi kutató nyert támogatást
Delmagyar.hu, a tanszékünk Lendület csoportjainak bemutatása (2021)
Az SZTE Fizikai Kémiai és Anyagtudományi Tanszéke a "legLENDÜLETesebb"
SZTE honlap, a tanszékünk Lendület csoportjainak bemutatása (2021)
"Bolyaisok" – Czakó Gábor elméleti kémikus
MTA honlap, a legsikeresebb Bolyai-ösztöndíjas kutatók bemutatása (2021)
Ismét díjazta kiváló oktatóit, kutatóit a Szegedi Tudományegyetem
SZTE honlap, az Év Fiatal Kutatója Elismerő Oklevélről (2020)
Kutatócsoportunk a Szeged TV műsorában
Szeged TV Kvantum (7:15-től), a Lendület pályázat elnyeréséről (2019)
Az energiahatékonyságot kutatják Szegeden: a nemzetközi élvonalba léphet Czakó Gáborék csoportja
Délmagyarország, a Lendület pályázat elnyeréséről (2019)
Gábor Czakó receives Momentum research grant (angolul)
Emory University honlap, a Lendület pályázat elnyeréséről (2019)
Az SZTE két kutatója, Czakó Gábor és Enyedy Éva Anna nyert 2019-ben az MTA Lendület programján
SZTE honlap, a Lendület pályázat elnyeréséről (2019)
A középkori zenei emlékektől a koszorúér-betegség kockázatbecsléséig – az MTA Lendület programjának új nyertesei
MTA honlap, a Lendület pályázat elnyeréséről (2019)
Elmélet és kísérlet együttműködése
Magyar Kémikusok Lapja, interjú a kutatómunkánkról (2019)
Publikálni és programozni
CAMPUS Plusz (eduline), a kutatócsoportunkról egy HVG kiadványban (2018)
Breaking the bond: To take part or not? (angolul)
ScienceDaily, a Science Advances cikkünkről (2018)
Elméleti kémiai és nemlineáris dinamikai kutatások a Szegedi Tudományegyetem Kémiai Intézetében
Magyar Kémikusok Lapja, bemutatkozik kutatócsoportunk (2018)
Czakó Gábor elméleti kémikus: "az az eredmény, amit a kutató nem publikál, nem is létezik"
SZTE honlap, egy újabb Nature Chemistry cikk megjelenése alkalmából (2017)
Még Amerikában is az SZTE-ről olvasnak!
SZEGEDMA online, egy összefoglaló cikk (J. Phys. Chem. A Feature Article) megjelenéséről (2017)
Study clarifies the role of the leaving group in gas-phase bimolecular reaction (angolul)
phys.org, a csoport első Nature Chemistry cikkéről (2015)
Szegedi kutatók eredményei a világ vezető kémiai folyóiratában
SZEGEDMA online, a csoport első Nature Chemistry cikkéről (2015)
Szegedi vegyészek úttörő felfedezése
Délmagyarország, a csoport első Nature Chemistry cikkéről (2015)
Szegedi kutatók eredményei a világ vezető kémiai folyóiratában
SZTE honlap, a csoport első Nature Chemistry cikkéről (2015)
Galéria